
概述
 腦膠質瘤
腦膠質瘤 腦膠質瘤病是一種罕見的中樞神經系統原發腫瘤。1938年Nevin首先提出。1943年由Scheinker和Evans命名為“瀰漫性腦神經膠質母細胞瘤病”,而前者則更優於後者,至今被許多文獻採納。以後根據其病理學的特點定義為神經膠質細胞瀰漫性瘤樣增生,而原有解剖結構保持完整為特徵的原發性腦瘤。
分類
常見分類有:星形細胞瘤、星形母細胞瘤、多形性膠質母細胞瘤、少枝膠質細胞瘤、室管膜瘤、脈絡叢乳頭狀瘤、松果體細胞瘤、神經元腫瘤及髓母細胞瘤。
類型
 腦膠質瘤
腦膠質瘤 腦膠質瘤有以下二種類型:
纖維型
是常見類型。腫瘤中有神經膠質纖維,這是與原漿型的主要區別,腫瘤質地較韌,瀰漫纖維型的切面呈白色,與腦白質不易區別。鄰近皮質常被腫瘤浸潤,色澤變深,與白質的分界模糊,腫瘤中心可有囊性變。局灶纖維型的邊界光整,主要見於小腦,常有囊性變。在鏡下間質中有神經膠質纖維,交叉分布於瘤細胞之間,瘤細胞為纖維型星形細胞。
原漿型
是最少見的一種類型。切面呈半透明均勻膠凍樣,深部侵入白質,邊界不清,常有變性,形成囊腫。在鏡下,腫瘤由原漿型星形細胞構成。
病因
膠質瘤起源於神經間膠質、室管膜、脈絡叢上皮、神經元等,起因至今未明。
特點
 腦膠質瘤
腦膠質瘤 臨床表現多種多樣而無特異性,神經影像學表現病變廣泛瀰漫,缺乏特徵性的跡象。本病有以下特點:
臨床特徵
亞急性起病,呈進行性發展趨勢。病程長短不一,患者平均生存期6~9個月,8.8%的病人能超過12個月。任何年齡都可以發病,在6~62歲之間起病較多。臨床缺乏特徵性局灶體徵,常以性格改變、精神異常、癲癇發作、顱內壓增高、偏癱為主要表現。JenninsMT等回顧85篇文獻報導的160例病人,44%~78%的患者有精神異常、智慧型減退,38%~50%有癲癇發作,39%~47%有顱內壓增高,58%有錐體束受損,37%有顱神經損害。實驗室檢查:腦脊液蛋白正常或輕度升高,白細胞數正常。腦電圖:瀰漫性慢波,偶見棘波。
神經影像學特點
病程呈瀰漫性浸潤生長,範圍廣泛,邊界不清,受累區域的腦組織腫脹,溝變淺或消失,腦室變小。病變早期占位效應常不明顯,中線結構常沒有移位。病變中、晚期可表現出占位效應,若病變偏一側,占位效應徵象可較早出現。腫瘤細胞多侵犯大腦半球2個或2個以上的部位,皮層及皮層下白質均可受累。
症狀
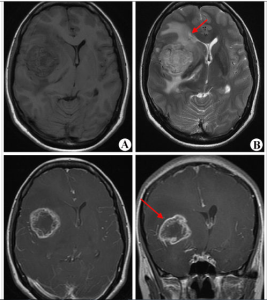 腦膠質瘤
腦膠質瘤 一是顱內壓增高和其他一般症狀,如頭痛、嘔吐、視力減退、復視、癲癇發作和精神症狀等。另一是腦組織受腫瘤的壓迫、浸潤、破壞所產生的局部症狀,造成神經功能缺失。
二、嘔吐:系由於延髓嘔吐中樞或迷走神經受刺激所致,可先無噁心,是噴射性。在兒童可由於顱縫分離頭痛不顯著,且因後顱窩腫瘤多見,故嘔吐較突出。
三、顱內壓增高:可產生視乳頭水腫,且久致視神經繼發萎縮,視力下降。腦神經膠質瘤壓迫視神經者產生原發性視神經萎縮,亦致視力下降。外展神經易受壓擠牽扯,常致麻痹,產生復視。
診斷
膠質瘤的診斷,根據其生物學特徵、年齡、性別、好發部位及臨床過程進行分析,在病史及體徵基礎上,採用電生理、超音波、放射性核素、放射學及核磁共振等輔助檢查,定位正確率幾乎是100%,定性診斷正確率可在90%以上。
預防
腦膠質瘤是最常見的顱內惡性腫瘤,由於其惡性程度、生長部位、治療手段與時機各不相同,故其預後亦不相同。早診治很重要。如有反覆頭痛、嘔吐等症狀,應想到本病可能,宜及早就醫。
經系統治療的患者,應面對現實,配合康復治療,定期複查;膠質瘤可復發,部分病人有多次手術機會。
