
流行病學
 腦內脊索瘤
腦內脊索瘤1962年Schisano報告6700例腦瘤中,僅有10例脊索瘤(0.15%)其他也有類似報告為0.2%以下,為少見的腫瘤。各家報告的年齡範圍為10~90歲(Hess,1934;Sassin,1967;Morello,1970)。發病年齡高峰為30~40歲,平均年齡為35~40歲之間。斜坡脊索瘤的臨床表現比骶尾部脊索瘤平均早10~15年男性比女性多見其比例為3∶2。在骶尾部腫瘤,有的病例組為2∶1或3∶1,而在顱內脊索瘤最接近的是1∶1。性別的優勢依然未能解釋,儘管推測外傷對腫瘤的產生可能起作用。
病因:
脊索瘤起源於胚胎殘留的脊索組織。在胚胎期間,脊索上端分布於顱底的蝶骨和枕骨,部分達到顱內面,並與蝶鞍上方的硬腦膜相銜接,在枕骨部分可達該骨之下面(即舌咽面),一部分亦可位於顱底骨和咽壁之間,脊索的下端分布於骶尾部的中央及中央旁等部位。當胎兒發育至3個月的時候脊索開始退化和消失,僅在椎間盤內殘留,即所謂的髓核。如果脊索的胚胎殘留在上述部位滯留到出生後可逐漸演變成腫瘤。因此脊索瘤好發於這些部位,尤以顱底蝶枕部和骶尾部為最多見,脊柱型者次之。
 腦內脊索瘤
腦內脊索瘤發病機制:
肉眼觀察腫瘤質地軟,呈膠凍狀,可有或無纖維包膜,早期與周圍腦組織的界限尚比較清楚,晚期則界限不清,浸潤破壞鄰近骨質和神經組織,引起顱底骨質的破壞腫瘤切面呈半透明,含有黏液樣物質,為腫瘤變性的產物,故其含量的多寡可以提示腫瘤的良惡與否。腫瘤中間有由包膜相連而形成的白色堅韌的間隔,將腫瘤分割成大小不等的多葉狀。半數瘤內有結節狀鈣化。腫瘤內可有出血和囊變。
鏡下可見典型的脊索瘤由上皮樣細胞所組成,細胞胞體大,多邊形,因胞質內含有大量空泡,可呈黏液染色故稱囊泡細胞或空泡細胞,細胞核小,分裂象少見,胞質內空泡有時合併後將細胞核推至一旁,故又稱為“印戒細胞”。有些地方細胞的界限消失,形成黏液狀合體。大量空泡細胞和黏液形成是本病的病理形態特點,近10%脊索瘤細胞增殖活躍,黏液顯著減少,並有核分裂現象細胞排列成條或島狀,埋於疏鬆的黏液組織之間,可含有軟骨組織、鈣化斑及小片骨組織。其周圍為網狀的結締組織所圍繞,將腫瘤分割成不規則小葉狀。
脊索瘤可分為:
1、普通型 又稱典型型,最常見。占總數80%~85%。瘤內無軟骨或其他間充質成分。多見於40~50歲患者,小於20歲者少見。無性別差異。在病理上可有幾種生長方式,但片狀生長為其特徵,由空泡狀上皮細胞和黏液基質組成。細胞角蛋白和上皮膜抗原的免疫染色陽性,電鏡見核粒,這些特徵有助於本病與軟骨肉瘤區別,後者免疫染色陰性,電鏡無核粒。
2、軟骨樣脊索瘤 占脊索瘤的5%~15%。其鏡下特點除上述典型所見外尚含有多少不等的透明軟骨樣區域。雖然有些作者通過電鏡觀察後將其歸類為低度惡性的軟骨肉瘤,但是大量的免疫組化研究卻發現軟骨樣脊索瘤的上皮性標記抗原呈陽性反應。本型發病年齡較輕,過去認為其預後普遍較普通型好,現在認為兩者預後差不多。
3、間質型 又稱非典型型,占脊索瘤的10%,含普通型成分和惡性間充質成分,鏡下表現為腫瘤增殖活躍,黏液含量顯著減少並可見到核分裂象。少數腫瘤可經血流轉移和蛛網膜下腔種植性播散。本型可繼發於普通型放療後或惡變。常在診斷後6~12個月死亡。
臨床表現
顱內脊索瘤為良性腫瘤,生長緩慢,病程較長,平均可在3年以上頭痛為最常見的症狀,約70%的病人有頭痛,有時在就醫前即已頭痛數年,常為全頭痛,也可向後枕部或頸部擴展。頭痛性質呈持續性鈍痛,一天中無顯著變化。如有顱內壓增高則勢必加重,脊索瘤的頭痛與緩慢持久的顱底骨浸潤有關,頭痛也可再發。
顱內脊索瘤的臨床表現可因腫瘤部位和腫瘤的發展方向而有所不同。
1、鞍部脊索瘤:垂體功能低下主要表現在陽萎、閉經、身體發胖等。視神經受壓產生原發性視神經萎縮,視力減退以及雙顳側偏盲等。
2、鞍旁部脊索瘤:主要表現在Ⅲ、Ⅳ、Ⅵ腦神經麻痹,其中,以外展受累較為多見,這可能因為展神經行程過長,另外,展神經的近端常是腫瘤的起源部位,以致其發生率較高。一般均潛在緩慢進展甚至,要經1~2年。腦神經麻痹可為雙側,但常為單側,難以理解的是往往在左側。
3、斜坡部脊索瘤:主要表現為腦幹受壓症狀,即步行障礙,錐體束征第Ⅵ、Ⅶ腦神經障礙,其中,雙側展神經損害為其特徵。
4、廣泛型:病變範圍廣泛,超出以上某一類型,甚至延伸至顱底移位區域,據有以上相關類型的臨床症狀和影像學表現。
併發症:
由於腫瘤發生於顱底,可引起交通性腦積水。如腫瘤向橋小腦角發展,則出現聽覺障礙,耳鳴、眩暈。脊索瘤起源於鼻咽壁近處,常突到鼻咽或浸潤一個或更多的鼻旁竇。引起鼻不能通氣、阻塞、疼痛,常見有膿性或血性鼻分泌物,也因機械性阻塞、致吞咽困難,鼻咽症狀常在神經受累之前出現,必須切記查看鼻咽腔有13%~33%的機會看到腫塊。
外科手術併發症包括腦脊液漏、腦膜炎和腦神經損傷(其中大多數能恢復)等。
診斷
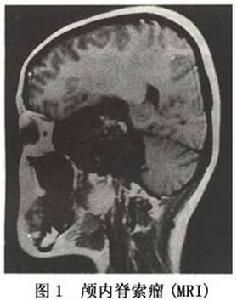 圖1 腦內脊索瘤
圖1 腦內脊索瘤成年患者有長期頭痛病史並出現一側展神經麻痹者,應考慮到脊索瘤的可能但確定診斷尚需藉助X線、CT和MRI等影像學檢查。
鑑別診斷:
脊索瘤應與腦膜瘤相鑑別。同部位腦膜瘤可引起局部骨質受壓變薄或骨質增生,而少有溶骨性變化。DSA常見腦膜供血動脈增粗,有明顯的腫瘤染色。
如脊索瘤向後顱窩生長應與橋小腦角的聽神經瘤作鑑別。聽神經瘤在顱骨平片和CT上主要表現為內聽道的擴大和岩骨嵴的吸收。MRI常有助於鑑別診斷。
鞍區部位的脊索瘤需與垂體腺瘤和顱咽管瘤相鑑別。後兩者多不引起廣泛的顱底骨質的破壞,垂體瘤在影像學上一般表現為蝶鞍受累擴大,鞍底變深,骨質吸收。顱咽管瘤CT上可見囊壁有弧線狀或蛋殼樣鈣化,通常不引起鄰近骨破壞,且兩者腦神經損害多局限於視神經,而脊索瘤多表現為以展神經障礙為主的多腦神經損害,影像學上多見顱底骨質溶骨性改變和瘤內斑點狀或片狀鈣化。
向下長入鼻咽部的脊索瘤因其臨床表現和X線檢查特徵與向顱底轉移的鼻咽癌相似,鑑別診斷主要依靠鼻咽部的穿刺活檢。
鞍旁型或長向中顱底的脊索瘤與軟骨肉瘤鑑別比較困難,免疫組化染色很有幫助,脊索瘤對多種組織標記物均顯示陽性,如:Cyto-K6/7EMA7/7、CEA6/7、GFAP0/7、Des0/7、α-AT7/7、Lyso4/7而軟骨肉瘤則均顯示為陰性。
其它輔助檢查:
1.頭顱X線平片:有重要診斷意義,表現為腫瘤所在部位的骨質破壞,也常見到斑片狀或團狀腫瘤鈣化。
2.CT掃描:可見顱底部類圓形或不規則略高密度影,邊界較清楚,瘤內有鈣化,增強掃描腫瘤不強化或輕度強化。骨窗像可見明顯骨質破壞。
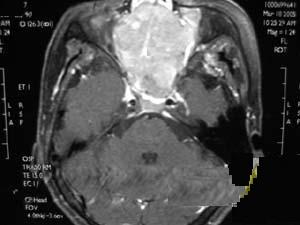
3.MRI:腫瘤多為長T1長T2信號,瘤內囊變區呈更長T1長T2信號,鈣化為黑色無信號影,出血灶則呈高信號注射Gd-DTPA後腫瘤輕度至中度強化(圖1)。
4.腦血管造影:如腫瘤位於鞍區,可見頸內動脈虹吸段外移,大腦前動脈水平段上抬。位於鞍旁顱中窩者,頸內動脈海綿竇段抬高,大腦中動脈起始段和水平段也上抬。向顱後窩發展的腫瘤常將基底動脈推向後方或側後方。
治療
治療原則
脊索瘤的傳統治療以手術切除為主,但正如Delgado等所指出的,顱底脊索瘤全部切除在技術上是不可能的,單純手術切除治療的局部腫瘤復發率高,高達85%,手術往往達不到全部切除的效果,因為:①腫瘤位於顱底,部位深在,無論採用何種手術進路常不能達到理想的暴露,視野受限。②腫瘤往往與重要神經(如視、面和米走神經等)、血管(如頸內、椎和基底動脈等)及腦和腦幹緊密相連,給手術帶來困難和危險,一旦損傷這些結構,容易引起嚴重後果,甚至招致死亡。
1、外科手術 由於次全切除腫瘤的5年生存率較活檢者長,因此應儘量切除腫瘤。許多學者仍致力於各種手術入路的選擇以求全切腫瘤,神經導航的套用也利於提高腫瘤全切率。但是,迄今沒有一種手術入路適用於全部脊索瘤,一些脊索瘤還需多種手術入路的聯合套用。在選擇手術入路時應考慮下列因素:腫瘤部位術者對各種可供選擇入路的掌握程度、手術組的經驗和配合、顱頸穩定性等。大多數脊索瘤位於硬膜外少數可破壞硬膜,長入蛛網膜下腔。因此,位居中線的脊索瘤可選用中線手術入路,如經口-硬齶入路、經蝶竇入路、擴大額下硬膜外入路、經上頜或經顏面入路等。偏側生長脊索瘤可用前外側硬膜外入路後外側(經髁)入路等。枕骨髁受累者不僅影響顱頸關節的穩定性,且術後易復發,在設計治療方案時要特別注意。
2、常規放射治療(包括γ-刀、和X-刀)
⑴體位及照射技術:病人一般採取臥位,斜架,頭墊合適角度的頭枕,使下頜儘量內收,保證前野垂直照射時避開眼球。採用面罩固定技術可也充分保證治療過程中的重複性和精確性。放療技術可用兩野(雙顳側野)對穿照射,但考慮到由於兩野對穿照射可造成顳葉受量過高,最好使用3野照射技術,即在2野的基礎上再加一個額部前正中野,3野等中心照射,既能使靶區得到滿意的劑量分布,又避免了雙側顳葉受量過高的副作用。具體技術類似於垂體瘤的3野照射技術。
⑵靶區設計:主張利用強化的CT或MRI影像所顯示的腫瘤大小及侵犯範圍而設計,照射邊緣根據具體腫瘤的大小外放0.5~1cm,並依靠TPS來精確制定放射治療計畫。
⑶能量和劑量:由於病變位於中線部位,因此主張高能X線照射,能量不低於6MVX線,有條件者可採用10MV、18MV X線照射。
採用常規分割照射技術,分次劑量1.8~2GY。因為腫瘤毗鄰重要機構如視神經、腦幹等,受安全劑量的限制,而且過高劑量的放療並不能進一步降低局部復發率,故放療總劑量一般在55~66GY(中值劑量60GY)
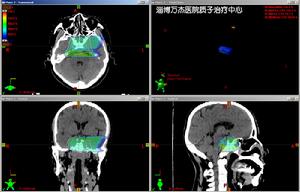 腦部脊索瘤質子治療計畫
腦部脊索瘤質子治療計畫3、質子照射技術
脊索瘤增殖緩慢,而且瘤體毗鄰重要機構如視神經、視交叉、腦幹等,因此保證瘤體得到較高劑量的照射、周圍重要結構處於相對安全的劑量,高線性能量傳遞放療設備如質子束有其套用價值。使用質子束進行照射有兩種方式,一種為單一質子照射,另外一種為質子與光子的混合照射。目前在臨床上多採用後一種治療方式。
在質子和光子的混合照射種,一般光子與質子的劑量比為4:1~2,有以下兩種混合方式:a、常規分割照射過程中,光子每周照射4次,第5次照射採用質子照射。如此,在總量
 腦部脊索瘤質子治療計畫
腦部脊索瘤質子治療計畫60CGE的前提下,光子照射的總量為50GY,而質子照射的總劑量為10CGE。b、先行光子照射40GY,然後採用質子束局部加量照射20CGE,使總量達到60CGE
Austin-Seymour總結194例術後接受質子束照射的脊索瘤和軟骨肉瘤病例,平均5年腫瘤復發控制率達76%,其中脊索瘤為64%。美國麻薩諸塞州總醫院(1995)報導204例脊索瘤病人,其中腫瘤位顱底153例,上頸椎51例。套用常規放療和質子刀聯合,平均劑量為70.1cGy(鈷灰當量),平均隨訪54個月(8~158個月),腫瘤總復發率31%,其中局部復發95%,遠隔復發20%,復發者3年生存率44%,5年生存率5%。預後良好者69%。放射外科治療預後不良與下列因素有關:①瘤容積>75ml;②瘤體壞死>10%;③頸椎同時受累;④女性。18例兒童(4~18歲)患者,平均劑量69cGy,隨訪72個月,5年生存率68%與成人療效相似
4、其他治療
包括熱療、90Y局部埋藏治療及化療等,但療效不肯定。
預後
預後因素
1、腫瘤體積和病變範圍腫瘤體積和腦幹受侵顯著影響局部控制率:腫瘤體積≤25ML者100%局部控制,而腫瘤>25ML的局部控制率僅為56%(P=0.02);無腦幹受侵者的局部控制率為94%,而腦幹受侵者的局部控制率為53%(P=0.04),組間比較均有顯著的統計學意義。
2、手術切除範圍
據Colli等報導53例顱底頸椎交接處脊索瘤的患者採用手術切除+術後放療,隨訪結果顯示,手術切除範圍顯著影響腫瘤的局部控制率:根治性切除和次全切的局部控制率明顯好於腫瘤部分切除。
3、年齡
Noel等於新近報導67例顱底和頸椎的脊索瘤、軟骨肉瘤(49例脊索瘤、18例軟骨肉瘤)採用光子和質子混合束照射的結果:2/3劑量採用光子束照射,1/3劑量採用質子束照射,總劑量60~70CGE,中位隨訪29個月(4~71個月),脊索瘤的3年控制率71%,而軟骨肉瘤為85%;3年總生存率前者為88%,後者為75%。14例在隨訪過程中出現局部復發(其中8例在GTV、4例在CTV、2例無法評價),局部復發率為21.5%。但因素統計學分析顯示年齡≤52歲、腫瘤體積<28ML顯著影響局部控制,而多因素分析僅年齡顯著影響預後。
單純手術組的局部復發率高達100%,復發的平均時間為2.5年,而術後放療可將復發率降至50%,而且復發的時間推遲至平均術後4年左右。
一般而言,綜合治療顱底脊索瘤總的5年生存率為33%~75%,10年生存率為17%~45%。
常規放療是外科治療的重要輔助手段,患者的生存率與放療劑量相關,Menezes的資料顯示:術後放療劑量<40GY,5年生存率為零。Forsyth報導了39例手術加常規放療的脊索瘤患者,5年生存率為51%,10年生存率為35%。
如術後放療採用質子束或質子束與光子束的混合照射,則可進一步改善顱底脊索瘤的預後。如Noel報導34例顱底脊索瘤採用光子和質子混合照射的結果:總劑量60~70CGE,GTV平均總劑量限制在67CGE,中位隨訪30.5個月(2~56個月),3年局部控制率為83.1%,總的生存率為91%,遠高於常規光子術後放療的局部控制率及生產率。
由於視神經、腦幹和脊髓等重要器官臨近顱底,因此常規光子治療無法予以高劑量照射,一般照射劑量不超過60GY,用質子照射時可把局部劑量提高到70~76CGE左右,脊索瘤10年控制率可達45%,HugEB等治療29例脊索瘤,放射治療劑量為50.4~78.6CGE,中位隨訪40個月(13~92個月),腫瘤局部控制率為60%。
| 治療方法 | 復發率 | 復發生存時間/年 | 備註 |
| 不治療 | 自然情況下1年死亡 | ||
| 單純手術 | 接近100% | 平均2.5復發 | |
| 手術加普通放療 | 降低50% | 復發推遲4年 | |
| 普通放療 | 5年生存率為0 | 劑量<40GY | |
| 質子+光子放療 | 3年控制率83.1% | 劑量60~70CGE | |
| 質子 | 10年控制率45% | 局部劑量70~76CGE |
