
定義
 腦脊索瘤
腦脊索瘤病因及機制
病因
脊索瘤起源於胚胎殘留的脊索組織。在胚胎期間,脊索上端分布於顱底的蝶骨和枕骨,部分達到顱內面,並與蝶鞍上方的硬腦膜相銜接,在枕骨部分可達該骨之下面(即舌咽面),一部分亦可位於顱底骨和咽壁之間,脊索的下端分布於骶尾部的中央及中央旁等部位。當胎兒發育至3個月的時候脊索開始退化和消失,僅在椎間盤內殘留,即所謂的髓核。如果脊索的胚胎殘留在上述部位滯留到出生後可逐漸演變成腫瘤。因此脊索瘤好發於這些部位,尤以顱底蝶枕部和骶尾部為最多見,脊柱型者次之。
發病機制
肉眼觀察腫瘤質地軟,呈膠凍狀,可有或無纖維包膜,早期與周圍腦組織的界限尚比較清楚,晚期則界限不清,浸潤破壞鄰近骨質和神經組織,引起顱底骨質的破壞腫瘤切面呈半透明,含有黏液樣物質,為腫瘤變性的產物,故其含量的多寡可以提示腫瘤的良惡與否。腫瘤中間有由包膜相連而形成的白色堅韌的間隔,將腫瘤分割成大小不等的多葉狀。半數瘤內有結節狀鈣化。腫瘤內可有出血和囊變。
鏡下可見典型的脊索瘤由上皮樣細胞所組成,細胞胞體大,多邊形,因胞質內含有大量空泡,可呈黏液染色故稱囊泡細胞或空泡細胞,細胞核小,分裂象少見,胞質內空泡有時合併後將細胞核推至一旁,故又稱為“印戒細胞”。有些地方細胞的界限消失,形成黏液狀合體。大量空泡細胞和黏液形成是本病的病理形態特點,近10%脊索瘤細胞增殖活躍,黏液顯著減少,並有核分裂現象細胞排列成條或島狀,埋於疏鬆的黏液組織之間,可含有軟骨組織、鈣化斑及小片骨組織。其周圍為網狀的結締組織所圍繞,將腫瘤分割成不規則小葉狀。
病理分型
普通型
又稱典型型,最常見。占總數80%~85%。瘤內無軟骨或其他間充質成分。多見於40~50歲患者,小於20歲者少見。無性別差異。在病理上可有幾種生長方式,但片狀生長為其特徵,由空泡狀上皮細胞和黏液基質組成。細胞角蛋白和上皮膜抗原的免疫染色陽性,電鏡見核粒,這些特徵有助於本病與軟骨肉瘤區別,後者免疫染色陰性,電鏡無核粒。
軟骨樣脊索瘤
軟骨樣脊索瘤占脊索瘤的5%~15%。其鏡下特點除上述典型所見外尚含有多少不等的透明軟骨樣區域。雖然有些作者通過電鏡觀察後將其歸類為低度惡性的軟骨肉瘤,但是大量的免疫組化研究卻發現軟骨樣脊索瘤的上皮性標記抗原呈陽性反應。本型發病年齡較輕,過去認為其預後普遍較普通型好,現在認為兩者預後差不多。
間質型
間質型又稱非典型型,占脊索瘤的10%,含普通型成分和惡性間充質成分,鏡下表現為腫瘤增殖活躍,黏液含量顯著減少並可見到核分裂象。少數腫瘤可經血流轉移和蛛網膜下腔種植性播散。本型可繼發於普通型放療後或惡變。常在診斷後6~12個月死亡。
症狀
顱內脊索瘤為良性腫瘤,生長緩慢,病程較長,平均可在3年以上頭痛為最常見的症狀,約70%的病人有頭痛,有時在就醫前即已頭痛數年,常為全頭痛,也可向後枕部或頸部擴展。頭痛性質呈持續性鈍痛,一天中無顯著變化。如有顱內壓增高則勢必加重,脊索瘤的頭痛與緩慢持久的顱底骨浸潤有關,頭痛也可再發。顱內脊索瘤的臨床表現可因腫瘤部位和腫瘤的發展方向而有所不同。
1.鞍部脊索瘤:垂體功能低下主要表現在陽痿、閉經、身體發胖等。視神經受壓產生原發性視神經萎縮,視力減退以及雙顳側偏盲等。
2.鞍旁部脊索瘤:主要表現在Ⅲ、Ⅳ、Ⅵ腦神經麻痹,其中,以外展受累較為多見,這可能因為展神經行程過長,另外,展神經的近端常是腫瘤的起源部位,以致其發生率較高。一般均潛在緩慢進展甚至,要經1~2年。腦神經麻痹可為雙側,但常為單側,難以理解的是往往在左側。
3.斜坡部脊索瘤:主要表現為腦幹受壓症狀,即步行障礙,錐體束征第Ⅵ、Ⅶ腦神經障礙,其中,雙側展神經損害為其特徵。
4、廣泛型:病變範圍廣泛,超出以上某一類型,甚至延伸至顱底移位區域,據有以上相關類型的臨床症狀和影像學表現。
併發症:
由於腫瘤發生於顱底,可引起交通性腦積水。如腫瘤向橋小腦角發展,則出現聽覺障礙,耳鳴、眩暈。脊索瘤起源於鼻咽壁近處,常突到鼻咽或浸潤一個或更多的鼻旁竇。引起鼻不能通氣、阻塞、疼痛,常見有膿性或血性鼻分泌物,也因機械性阻塞、致吞咽困難,鼻咽症狀常在神經受累之前出現,必須切記查看鼻咽腔有13%~33%的機會看到腫塊。外科手術併發症包括腦脊液漏、腦膜炎和腦神經損傷(其中大多數能恢復)等。
診斷
成年患者有長期頭痛病史並出現一側展神經麻痹者,應考慮到脊索瘤的可能但確定診斷尚需藉助X線、CT和MRI等影像學檢查。鑑別診斷
脊索瘤應與腦膜瘤相鑑別。同部位腦膜瘤可引起局部骨質受壓變薄或骨質增生,而少有溶骨性變化。DSA常見腦膜供血動脈增粗,有明顯的腫瘤染色。如脊索瘤向後顱窩生長應與橋小腦角的聽神經瘤作鑑別。聽神經瘤在顱骨平片和CT上主要表現為內聽道的擴大和岩骨嵴的吸收。MRI常有助於鑑別診斷。
鞍區部位的脊索瘤需與垂體腺瘤和顱咽管瘤相鑑別。後兩者多不引起廣泛的顱底骨質的破壞,垂體瘤在影像學上一般表現為蝶鞍受累擴大,鞍底變深,骨質吸收。顱咽管瘤CT上可見囊壁有弧線狀或蛋殼樣鈣化,通常不引起鄰近骨破壞,且兩者腦神經損害多局限於視神經,而脊索瘤多表現為以展神經障礙為主的多腦神經損害,影像學上多見顱底骨質溶骨性改變和瘤內斑點狀或片狀鈣化。
向下長入鼻咽部的脊索瘤因其臨床表現和X線檢查特徵與向顱底轉移的鼻咽癌相似,鑑別診斷主要依靠鼻咽部的穿刺活檢。
鞍旁型或長向中顱底的脊索瘤與軟骨肉瘤鑑別比較困難,免疫組化染色很有幫助,脊索瘤對多種組織標記物均顯示陽性,如:Cyto-K6/7EMA7/7、CEA6/7、GFAP0/7、Des0/7、α-AT7/7、Lyso4/7而軟骨肉瘤則均顯示為陰性。
其它輔助檢查
1.頭顱X線平片有重要診斷意義,表現為腫瘤所在部位的骨質破壞,也常見到斑片狀或團狀腫瘤鈣化。2.CT掃描可見顱底部類圓形或不規則略高密度影,邊界較清楚,瘤內有鈣化,增強掃描腫瘤不強化或輕度強化。骨窗像可見明顯骨質破壞。
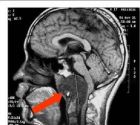
3.MRI腫瘤多為長T1長T2信號,瘤內囊變區呈更長T1長T2信號,鈣化為黑色無信號影,出血灶則呈高信號注射Gd-dtpa後腫瘤輕度至中度強化(圖1)。
4.腦血管造影如腫瘤位於鞍區,可見頸內動脈虹吸段外移,大腦前動脈水平段上抬。位於鞍旁顱中窩者,頸內動脈海綿竇段抬高,大腦中動脈起始段和水平段也上抬。向顱後窩發展的腫瘤常將基底動脈推向後方或側後方。
