
簡介
 四氫生物蝶呤缺乏症
四氫生物蝶呤缺乏症四氫生物蝶呤(BH4)缺乏症(tetrahydro biopterin deficiency),是高苯丙氨酸血症的一種亞型,是一種常染色體遺傳性疾病,與苯丙酮尿症(PKU)同屬於高苯丙氨酸血症(HPA),是截至21世紀初為止已確認的5000~6000種人類的罕見遺傳代謝病之一,主要會對人的神經系統造成損害,導致患兒出現智力低下、癲癇等症狀。
四氫生物蝶呤缺乏症通過對新生兒進行疾病篩查,檢出高苯丙氨酸血症後可得到進一步確診。在中國大陸地區,新生兒高苯丙氨酸血症的發病率約為1.1萬分之一;而在高苯丙氨酸血症中約有近兩成是BH4缺乏症。按這個發病率計算,在中國每年約2000萬新生兒中可能患BH4缺乏症的為100~200人。由於BH4缺乏症分經典型和非經典型兩種,因此早期很容易誤診。
病因
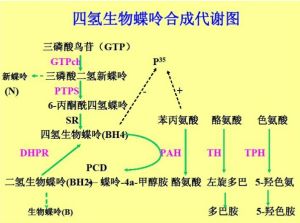 bh4合成代謝圖
bh4合成代謝圖已報導的BH4缺乏症有GTP環羥化酶Ⅰ(GTP cyclohydrolaseⅠ,GPTCH)、6-丙酮醯四氫蝶呤合成酶(6-pyruvoyltetrahydropterinsynthase,PTPS)、二氫蝶啶還原酶(dihydropteridinereductase,DHPR)和蝶呤-4a-甲醇胺脫水酶(pterin-4a-carbincl amine dehydratase,PCD)四個酶的異常。
其遺傳形式均為常染色體隱性遺傳。
臨床表現
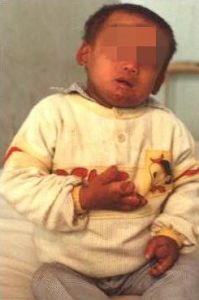
BH4缺乏症的新生兒出生時大多表現正常,除了血苯丙氨酸增高外,無任何臨床表現,往往被誤認為經典型PKU而給予低血苯丙氨酸飲食治療。
患兒出生3~4月後出現神經系統症狀,並逐漸出現比如特殊體味(全身和尿液有特殊鼠臭味);頭髮由黑變黃,虹膜顏色變淺,皮膚白,常伴有濕疹;精神、運動發育落後。
5個月時出現肌張力低下、痙攣和四肢鉛管狀僵硬等症狀,隨著年齡的增長,智力低下顯著,肌張力異常,或常常出現行為異常、興奮不安、多動、攻擊性行為,約有兩成患兒伴有癲癇發作等症狀。
中樞神經系統症狀較PKU嚴重且伴有難以糾正的酸中毒為本症的特點。
診斷
篩查尿篩查
與PKU患者相似,尿中出現PA、mandelate、PLA、2HPA、Phe、PPA、4HPL明顯增高。
依據上述尿篩查結果和血中BH4水平低下,同時伴有血中、尿中5-羥基吲哚乙酸(5-OH-indol acetate)、高香草酸(Homovanillicacid)、香草扁桃酸(vanilly mandelate)低下與PKU不同,是本病診斷的重要參考依據。
此病給於BH4治療有明顯的效果為鑑別診斷時的重要參考依據。
酶活性測定
確診需要做酶活性測定。
治療
經診斷為四氫生物蝶呤缺乏症的患者,應給予四氫生物蝶呤以降低血中苯丙氨酸濃度,還應根據具體情況在醫生的指導下補充5-羥色胺和美多巴(因BH4很難通過血腦屏障,單獨給與BH4很難預防神經系統症狀的發生),以維持腦和神經肌肉功能正常。必要時還可以考慮同時合用低苯丙氨酸飲食治療。
預後
BH4缺乏症患兒如果能夠在早期得到正確治療,可以像常人一樣健康生活。一般情況下,BH4缺乏症患兒在給予聯合藥物口服治療後,臨床症狀會好轉,尤其是3個月內及時治療的患兒,其智力發育接近正常兒童。
