
病因
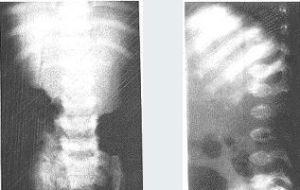 假性軟骨發育不良
假性軟骨發育不良另外有相當罕見的情況,未罹病的父母在精卵受精時有可能會產生性腺鑲嵌型(gonadalmosaicism),所以同時會產生正常和異常的精卵,當發生這樣的情形後,下一胎髮生性腺鑲嵌型(gonadalmosaicism)的再發率則會增加4%的遺傳風險。
軟骨低聚體基質蛋白基因(Cartilageoligomericmatrixproteingene,COMP)也已經被鑑別出可導致多發性骨發育不全症MultipleEpiphysealDysplasia(MED),意味著一些多發性骨發育不全症(MultipleEpiphysealDysplasia,MED)的類型,有假性軟骨發育不全(Psedoachondroplasia)的等位基因(allele)。
症狀
生長發育
身體肢體較短為一種生長的缺陷,發生於出生後。可於18個月大時至2歲之間被觀察到。
成人期的身高為82-130公分。
顱顏部
頭圍與臉部正常。
肢體
比較短且呈不成比例分布,韌帶鬆弛與關節過度伸展,尤其是手部,膝部與腳踝等部位。主要部位關節(除了肘部以外)會過度變形顯著(hypermobile),膝外翻(genuvalgum)與膝內翻(genuvarum)以及反屈(recurvatum)等症狀。手部尺側彎曲(ulnardeviation),手指較短且過度變形顯著(hypermobile)。手肘與髖部伸展受限制,下肢肢體畸形,輕微脊柱側彎,脊柱前凸(約50%的患者),兒童時期患有關節痛尤其是在下肢末端較大關節處。
影像檢查
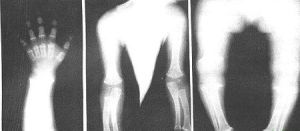 假性軟骨發育不良
假性軟骨發育不良顯著的短指畸形(brachydactyly),掌骨與指骨過短,小而不規則的腕骨(carpal)。
脊柱的異常包括扁平的角度變異性並伴隨有雙凹終板,以及來自於身體體表前端的中央前端骨頭的破裂。而腰椎上下的椎弓根寬度距離是正常的。
有齒突骨發育不全(odontoidhypoplasia)的現象,薦骨切跡(sacralnotch)較短。肋骨傾向變成匙型,終端的趾骨也較小。
腰椎脊柱前凸與後凸,脊柱側彎。
診斷
X線改變有一定特徵性,且為診斷及鑑別的主要依據.本病應與軟骨發育不全、粘多糖Ⅳ型、多發性骨骺發育不良、脊柱骨骺發育不全(先天型)等相區別。
治療
臨床處理以受侵犯小兒骨科合併症為主,關節痛可以止痛劑來控制,但並無系統性研究可評估在此症中疼痛控制的多變類型有無其有效作用。在兒童時期通常會進行切骨術來減低肢體的畸形現象。
需要接受手術治療脊柱側彎的現象並不常見,但是針對嚴重脊柱側彎的患者時就必需考慮採用手術來進行矯正。有關身材短小所帶來的心理衝擊,包括污名化(stigmatization)與歧視(discrimination)等狀況,必需特別小心處理,並且給予社會支持。
針對身體上的主要臨床特徵給予預防處理,當患者活動時應保護關節避免受到傷害。
