
概述
丹麥技術大學的J.Ruzicka 和E.H.Hansen於1975年提出了流動注射分析(Flow Injection Analysis,FIA)的新概念。把試樣溶液直接以“試樣塞”的形式注入到管道的試劑載流中,不需反應進行完全,就可以進行檢測。擺脫了傳統的必須在穩態條件下操作的觀念,提出化學分析可在非平衡的動態條件下進行,從而大大提高了分析速度。
 全自動流動注射分析儀
全自動流動注射分析儀流動注射分析技術在常規體積樣品預處理的自動化、微型化和線上化方面引起了革命性的變化,不僅極大地提高了整個分析過程的效率、可靠性和分析速度,減少了樣品的污染,也降低了樣品及試劑的消耗和廢液產量。更重要的是使某些難以或無法實現的手工操作成為可能且十分有效。自1975年引入流動注射分析技術以來,其在自動分析和聯用技術中已具有不可替代的重要作用。該領域中出版的有關專著已達20餘部,發表的研究論文已超過13800篇。從如此大量的文獻中可以清楚地看出流動注射分析技術發展的脈絡,或其在發展過程中經歷了第一代流動注射,第二代順序注射和第三代順序注射-“閥上實驗室”。
基於微型填充柱的線上固相萃取分離富集技術與原子光譜的聯用受到了廣泛的關注。在常規流動注射/順注射微填充柱分離富集體系中,微柱通常被視為整個體系的一個固定單元。柱子的容量、吸附劑的顆粒尺寸、溶脹性以及基體或共存組份的干擾程度等因素對於分析過程的重現性和可靠性均具有重要影響。一般而言,較小的顆粒尺寸有利於增加微型柱的容量,但經多次吸附-淋洗操作後,較小的吸附劑顆粒傾向於被逐漸壓緊而導致產生流動阻力;另一方面,對於由溶脹性比較顯著的吸附劑裝填的微型柱,如chelex100螯合樹脂,由於樹脂顆粒的溶脹而產生的壓力可能使液流無法流過微型柱而導致整個系統失效。微柱逆流淋洗法和微量空氣倒吸法對於緩和或減小微柱反壓力的影響具有一定作用,但若要有效消除反壓力則是十分困難的。另外,經反覆樣品吸附-淋洗操作後,吸附劑顆粒表面的污染以及有效功能團或活性位的損失等均很大程度地導致柱效的降低。Ruzicka等提出的可更新表面技術為解決上述問題提供了十分有效的途徑,其基本思想是在每一輪分析完成後將用過的吸附劑排入廢液,而為下一輪分析裝填新柱。這一技術可以徹底消除由於吸附劑表面性能的不可逆變化而引起的柱效降低,並解決由於固定柱重複使用而產生的流動反壓所導致的分析結果不可靠等問題。在“閥上實驗室”中引入可更新表面微型填充柱不僅避免了常規線上填充柱固相萃取分離富集所面臨的上述困難,而且超微型化可更新填充柱的使用及“閥上實驗室”的集成化流控特徵為分離富集體系的進一步微型化開闢了新的途徑。這是基於微珠注射的“閥上實驗室”系統。
基本原理
Ruzicka等1988年在其專著第二版中對流動注射分析作的定義為:向流路中注入一個明確的流體帶,在連續非隔斷載流中分散而形成濃度梯度,從此濃度梯度中獲得信息的技術。
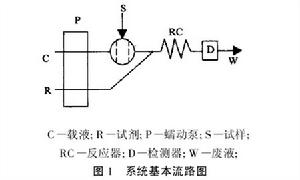
FIA基本流路系統一般包括:載液驅動系統、注入閥或進樣器、微型反應器、流通式檢測器(折光計、比色計、紫外/可見分光光度計、離子選擇電極、原子吸光光度計、螢光計等)、信號記錄裝置。其中蠕動泵驅動載液以恆定流率流過細微的管路;注入閥將一定體積的樣品溶液重現地注入載液中;微型反應器則使注入的樣品帶在其中適當地分散,並與載液(或試劑)中某些組分進行反應,生成能使檢測器產生適量回響值的產物;檢測器和信號記錄裝置測量和記錄下回響值數據。右圖是FIA的基本流路圖。反應器中的反應可以是不完全反應,只要其中的分散和反應可以高度重現就可以,而FIA體系恰好能滿足重現性良好的要求。並且FIA能控制試樣的分散,從而能有效控制樣品的稀釋度,進而縮短反應時間,提高檢測效率,所以FIA已成為線上檢測的理想工具。
試樣和試劑的分散是所有FIA方法的核心問題,通常用分散係數D來描述試樣的分散狀態。D的定義是:決定分析讀數的流體微元組分在擴散過程發生前(C0)與發生後(Cmax)的濃度比值,即D=C0/Cmax。FIA體系中的分散過程是許多不同因素(包括流速、管道長度、管徑、試樣體積與檢測方式等)的複雜函式。主要影響有:①試樣的進樣體積越大,D越小。②反應器管長度越大,D越大。③管路集合形狀越複雜,試樣在其中流動方向改變越多,D越大。如:直管反應器的D最小,盤管與編織管(3D)反應器的D較大。④流速對D的影響與反應器的管徑大小有關,關係較為複雜。
主要特點
1、經濟性:快速開關和結果顯示,分析速度快,一般可達每小時進樣100-300次。從樣品注入到檢測器回響的時間間隔一般小於1min。
2、預製的方法盒使操作簡便,快捷。
3、數字矩陣檢測器可以削弱折射指數和氣泡的影響。
4、可至多同時3個參數。
5、1-3通道自由組合。
儀器組成
典型的FIA儀是由以下幾部分組成:
1、泵:用於驅動載流通過細管。
2、採樣閥:可重現地將一定體積樣溶液注入載流。
3、微型反應器:樣本帶在其中分散並與載流中的組分反應,成為流通檢測器所回響的產物。
4、檢測器:檢測流體的吸光度、電極電位或其它物理特性並記錄。
套用
 流動注射分析
流動注射分析鑒於其操作的方便性和微通道設計的多樣性,可以預期這種流控技術將在生命科學分析和複雜基體樣品超微金屬的分離富集中得到廣泛套用。在超微分離方面,主要套用尚局限於閥內超微型填充柱固相萃取分離,聯用的檢測器也僅為ETAAS和ICPMS。實際上,SI-LOV流控系統可與各種檢測器聯用,尤其適合於與微量連續進樣檢測器結合。目前所見的主要分離、富集方式均可在該系統中進行超微分離富集操作,包括閥內液-液萃取,閥內微滲析,沉澱/(共)沉澱及氫化物發生等。另外,SI-LOV的流控特徵使其十分適合於在生命科學分析中套用,包括閥上酶聯免疫分析、生命代謝過程中的無損原位分析、活體分析和單細胞分析等。將分析所需要的檢測器集成在閥上,則可實現真正意義上的“閥上實驗室”分析。在分析儀器的微型化中,SI-LOV還將是對晶片實驗室(lab-on-a-chip)或微全分析系統(μTAS)有關技術平台的重要補充。巨觀試樣的引入與前處理仍然是μTAS發展中的瓶頸和薄弱環節。這主要源於巨觀處理技術(包括傳統的流動注射分析系統)與μTAS在樣品和試劑處理規模上相差五、六個數量級(前者多為0.01~1mL,而後者常僅為1~100nL水平)。由於SI-LOV可有效地進行微升水平的液流流控,因此可能成為μTAS解決試樣引入與處理的理想手段,並成為其重要的組成部分。為區別於μTAS中的核心技術——— 微流控分析系統認為:明確提出以LOV為核心,在0.1~10(100)μL水平上發展介觀流控分析系統)將進一步促進這一介觀分析領域的發展並最終促進分析系統的微型化及其在生命科學中的套用。
故障來源
在實驗中,可能遇到三種來源的故障,一種是源於試樣材料的性質;另一種源於流動注射分析設備性能不佳;第三種是源於化學過程的設計欠妥。
在把試樣注入到流動注射分析體系之前,有時可能需要進行某種前處理,如稀釋、中和、過濾等,即使是高濃度、高酸(鹼)度或高粘度的試樣,也可以通過把數微升試樣注入到匯合式流路中直接進行稀釋。如果一開始就有懸浮固體,過濾當然是不可避免的,但是分析過程中也可能形成沉澱,固體顆粒不僅可能堵塞管路,也同樣可能對感測器產生干擾,尤其是在光學體系中。通常可以經過濾或離心來保證試樣的清潔,但防止由於化學反應而產生的固體顆粒就更要困難些,然而可以通過加入適當的的表面活性劑雙洗滌劑來防止生成沉澱物。
儀器故障分析

儀器故障可以通過記錄的峰形進行診斷。這可以在進行化學分析或分散係數測定的過程中來觀察:
1、重複進樣時重現性不佳:先檢查攜出,這可很容易地通過交替注入高濃度與低濃度的試樣來完成。消除攜出的方法是降低採樣頻率或增載入流泵速,也可以同時採取這兩項措施。還要檢查一下閥是否有漏泄,如果用自動的流動注射分析體系,還應檢查一下試液杯中的試液是否充足。
2、記錄峰在返回基線的過程中回響遲鈍。體系中死體積過大(接頭不良,流通池體積或其接口處體積太大),起到一個或若干個小“混合室”的作用,必須縮小或消除這些死體積。
3、基線漂移:在光學檢測系統中,其原因可能某些物質在流通池窗口上的澱積,通常可注入一種能溶解該澱積物的試劑來除去,也可用適當的洗液或洗滌劑沖洗整個系統,在電位檢測系統中,漂移可能來自標準電池電位的變化(可能為指示電極或(和)參比電極的E0值漂移的結果)也可能來自接界電位的變化。
4、氣泡。載流液未經脫氣處理,化學反應產生氣體(如CO2生成節)或由於在流通池中產生突然壓降(流通地接頭內徑大於接入管道的內徑得起Venturj效應所致。欲消除此種效應可把連結管深插到接頭中,使管口儘可能接近池腔,並在流通池的出口處連線一段長20cm,內徑0.5mm的管路,讓廢液經此管流向下一段廢液管。
5、峰上突發噪聲:檢測器中有小氣泡流過,出現氣泡可能是由於上面已提到的各種原因,也可能是採樣閥試樣孔(或環)未能全部充滿所致(檢查一下採樣閥)
6、信號噪聲顯著,基線雖然穩定,但記錄筆的動作不太平滑,記錄峰呈鋸齒狀。泵的脈動過大,可能是泵結構的缺點.也可能是泵管壓得不夠緊,還可能是泵管已舊,應當更新(當不工作時.應該鬆開泵管以延長其使用壽命)如果使用的是電位流通池.則噪聲可能來自靜電,欲消除這種噪聲,可在緊靠滾槓的泵管兩端各插入一小段金屬管,並將兩者短路後接地。
7、雙峰。由試樣和試劑混合不完全造成的試劑不足所引起。雖然在多數情況下觀察到的是峰上出現的噪聲,但在極端的情況下也可遇到雙峰,增加留存時間(降低泵速),增強混合(用匯合法),或減少試樣體積都可以消除這種現象。
8、在低濃度是峰的重現性良好但在高濃度時變壞。造成此現象的原因是在試樣濃度高的情況下試劑不足。糾正的方法是增大試劑的濃度,或將濃度較高的試樣稀釋,亦可同時採用這兩項措施。
9、負峰。當載流溶液有色而被注人的試樣無色且濃度低時,可造成載流液的局部稀釋而產生負峰。當試樣
與載流的粘度或化學組成差異較大時也會產生這種現象。採用基體和試樣組成類似的溶液作載流可有效地消除這種基體效應。先把試樣注入到惰性載流中,然後再與試劑匯合。
最後還應指出:當使用有電機控制的閥來進行自動注入時,如果能在一段選定的時間內使注入閥從注入位回復到充樣位,則可以用來減少峰寬從而擴大採樣頻率。可以讓閥在注入位停足夠長的時間或使定量孔中的試樣全部排出,但也可令其在注入位停留較短的時間,而僅排出部分試樣,不過如果注入時間過短,則可能使峰高降低,重現性變差。
