成份
活性成份:雷珠單抗
化學名稱:G1,抗-(人血管內皮生長因子)Fab片段(人-鼠單克隆rhuFabV2γ-1鏈),二硫鍵結合人-鼠單克隆rhuFabV2κ-鏈)
分子量:48KD
【處方組成】
每1ml含10mg雷珠單抗。
本品所含輔料為:α,α-海藻糖二水合物;組氨酸;鹽酸組氨酸一水合物;聚山梨醇酯20。
性狀
透明至微乳白色液體。
適應症
用於治療濕性(新生血管性)年齡相關性黃斑變性(AMD)。
規格
10mg/ml,每瓶裝量0.2ml。
用法用量
本品應在有資質的醫院和眼科醫生中使用。醫院應具備該疾病診斷和治療所需的相關儀器設備和條件,眼科醫生應具備確診濕性年齡相關性黃斑變性的能力和豐富的玻璃體內注射經驗。
本品經玻璃體內注射給藥。推薦劑量為每次0.5mg(相當於0.05ml的注射量),每月一次給藥。
如果不能長期每月注射給藥,也可在初始3個月連續每月注射1次給藥之後,按每3個月注射給藥1次。與持續每月注射相比,在初始3個月,連續每月注射之後的9個月治療中,如果按每3個月給藥1次,則視力改善將平均減少約5個字母(ETDRS視力或Snellen視力表1行)。
治療期間應每月監測患者視力變化情況,如果出現顯著的視力下降,需進一步接受本品注射治療。兩次注射之間的間隔時間不得小於一個月。
給藥方法:
在玻璃體內注射給藥前,應對患者的既往病史進行全面的評估,以評估其發生高敏反應的可能性。(見【警告】與【注意事項】)
本品必須在無菌條件下進行玻璃體內注射,其中包括採用外科手術的手部消毒、無菌口罩、無菌手套、無菌手術單和無菌開瞼器(或類似器具)。注射前必須給予患者適當的麻醉劑和眼局部用光譜抗生素。注射前消毒眼周皮膚、眼瞼和眼球表面。
應指導患者在每次注射前後3天自行滴注抗生素滴眼液,每天4次。
採用無菌技術,通過與1ml無菌注射器相連的18G(5μm)濾過針頭抽取本品瓶內的所有(0.2ml)內容物。濾過針頭不得用於玻璃體內注射,抽取內容物後必須丟棄。濾過針頭必須替換為無菌30G針頭,用於玻璃體內注射。必須排空注射器內空氣,直至注射器內芯尖端對準注射器上0.05ml的刻度線。
注射針頭應於角鞏膜緣後3.5~4.0mm處,對準眼球中心,向玻璃體內進針,避免水平進針。
緩慢推送0.05ml注射液,應注意在之後的注射時改變鞏膜注射部位。
注射後必須監測患者的眼內壓和眼內炎。監測應包括注射後立即檢查視神經乳頭的血流灌流、30分鐘內測眼內壓及2~7天后進行檢眼鏡、裂隙燈和眼底檢查。需指導患者立即向其醫生報告任何出現的眼內炎的症狀(見【注意事項】)。
每瓶注射液僅用於治療一隻眼的單次注射。如果對側眼也需要治療,必須使用新的一瓶注射液,並在向另一隻眼注射本品前更換無菌區、注射器、手套、手術單、開瞼器、濾過針頭和注射針頭。
特殊人群用藥
腎損害
腎損害患者無需調整用藥劑量(見【藥代動力學】)。
肝損害
尚無相關研究。由於全身暴露可忽略不計,無須採取特別措施。
老年患者
無需調整劑量。
不良反應
在三項Ⅲ期臨床試驗中共有1315名患者組成了安全性人群。所有患者接受至少24個月的本品治療。440名患者接受了0.5mg的推薦劑量的治療。
以下嚴重不良事件與注射操作有關:眼內炎、孔源性視網膜脫離、視網膜撕裂和醫源性外傷性白內障(見【注意事項】)。
在接受本品治療的患者觀察到的其他嚴重眼部不良事件包括眼內炎症與眼內壓升高(參見【注意事項】)。
在三項對照的Ⅲ期試驗FVF2598g(MARINA)、FVF2587g(ANCHOR)和FVF3192(PIER)合併的數據中,以下列出的不良事件在0.5mg本品治療組中的發生率(至少高出2個百分點)高於對照組(假注射治療或維替泊芬光動力療法[PDT])。因此這些被認為是潛在的藥物不良反應。下文提供的安全性數據還包括所有440例接受0.5mg本品的合併患者人群中發生的至少與注射本身或醫藥產品可能有關的不良事件。
不良反應按系統器官類別和頻率列出,頻率使用以下規定:很常見(>1/10),常見(>1/100至10),不常見(>1/1000至100),罕見(>1/10000至<1/1000),非常罕見(<1/10000)。
 雷珠單抗注射液
雷珠單抗注射液 雷珠單抗注射液
雷珠單抗注射液免疫原性:
與所有治療性蛋白相似,在接受本品治療的患者中可能出現免疫應答。免疫原性數據反映了在免疫試驗中,試驗結果提示本品抗體陽性的患者百分比,並高度依賴於試驗的靈敏度和特異性。
在不同治療組中,本品免疫反應性的治療前發生率為0%~5%。每月注射本品,為期6至24個月後,大約1%至8%的患者中可檢出本品抗體。
目前,對本品的免疫反應性的臨床顯著意義尚且不明。在免疫反應性水平最高的新生血管性年齡相關的黃斑變性患者中,部分可出現虹膜炎或玻璃體炎。
禁忌
對本品或本品成份中任何一種輔料過敏者禁用。
活動的或懷疑的眼部或眼周感染的患者。
活動期眼內炎症的患者。
注意事項
玻璃體內注射,包括本品注射,與眼內炎、眼內感染、孔源性視網膜脫離、視網膜撕裂和醫源性外傷性白內障有關(參見【不良反應】)。本品注射時必須採用合格的無菌注射技術。此外,注射後一周內應監測患者的情況,從而早期發現感染並治療。應指導患者在出現任何提示有眼內炎的症狀或任何上述提到的事件時,應立即報告給醫生。
本品注射後60分鐘內可觀察到眼內壓升高(參見【不良反應】)。因此須同時對眼內壓和視神經乳頭的血流灌注進行監測和適當治療。
玻璃體內使用血管內皮生長因子(VEGF)抑制劑後,存在潛在的動脈血栓栓塞事件的風險。在臨床Ⅲ期研究中,動脈血栓栓塞事件的發生率在本品和對照組之間是相近的。接受本品0.5mg的患者與本品0.3mg或對照相比,卒中的發生率在數值上較高,不過此差異並無統計學顯著性。卒中率的差異在具有已知卒中風險因子的患者,包括既往卒中病史或短暫性腦缺血發作史的患者中更大。因此主治醫生應對這些患者謹慎評價本品治療是否合適,以及治療益處是否超過了潛在的風險。
在所有治療用蛋白質藥物一樣,本品有潛在的免疫原性。
尚未研究雙眼同時使用本品治療的安全性與有效性。如果雙眼同時接受治療,可能會使全身暴露量升高,從而導致全身不良事件的風險升高。
本品不得與其他抗血管內皮生長因子(VEGF)藥物同時使用(全身或局部使用)。
出現下述情況,應暫停給藥,且不得在下次計畫給藥時間之前恢復給藥:
與上次的視力檢查相比,最佳矯正視力(BCVA)的下降≥30字母;
眼內壓≥30mmHg;
視網膜撕裂;
涉及中心凹中央的視網膜下出血,或出血面積占病灶面積的50%或更多;
在給藥前後的28天已接受或計畫接受眼內手術。
接受抗-VEGF治療濕性AMD之後,視網膜色素上皮撕裂的風險因素包括大面積的和/或高度隆起的視網膜色素上皮脫離。在具有這些視網膜色素上皮撕裂風險因素的患者中開始本品治療時應謹慎。
在孔源性視網膜脫離或3或4級黃斑裂孔患者中應中斷治療。
本品治療可引起短暫的視覺障礙,這可能影響駕駛或機械操作的能力(參見【不良反應】)。出現這些症狀的患者在這些暫時性的視覺障礙副作用消退前不能駕駛或進行機械操作。
孕婦及哺乳期婦女用藥
妊娠
目前尚無本品在妊娠婦女中使用的數據。未進行本品的動物生殖研究。同樣尚不清楚妊娠婦女使用本品是否會對胎兒造成傷害,或者會影響生育能力。本品不得用於妊娠婦女,除非預期利益超過對於胎兒的潛在風險時才可考慮使用。
有生育力的婦女
有生育能力的婦女應在治療期間採取有效的避孕措施。
哺乳
不清楚本品是否分泌入人乳汁中。作為預防性措施,建議患者在本品治療期間不要哺乳。
兒童用藥
由於缺乏此亞組人群的安全與有效性數據,因此不建議兒童與青少年使用本品。
老年用藥
在臨床試驗中,大約82%(1146/1406)的隨機接受本品治療的患者年齡≥65歲,大約55%(772/1406)的患者年齡≥75歲。在這些試驗中,隨著年齡增加,本品的有效性或安全性未出現顯著差異。在人群藥代動力學分析中,經過肌酐消除率校正後,年齡對於全身暴露水平不存在顯著影響。
藥物相互作用
目前尚未進行正式的藥物相互作用研究。
藥物過量
臨床試驗與上市後數據中已報告了意外用藥過量的病例。與這些報告的病例最常相關的不良事件有眼內壓升高和眼痛。如果出現藥物過量,應監測眼內壓並治療(如果主治醫生認為有必要時)。
臨床試驗
藥效動力學性質
本品是一種人源化的重組單克隆抗體片段(Fab),靶向抑制人血管內皮生長因子A(VEGF-A)。它與VEGG-A亞型(即VEGF110、VEGF121、VEGF165)以較高的親和力,從而抑制了VEGF-A與其受體VEGFR-1和VEGFR-2的結合。VEGFA與其受體結合,可導致血管內皮細胞增殖和新生血管形成,以及增加血管滲漏,所有這些被認為與新生血管性年齡相關性黃斑變性(AMD)的進展相關。
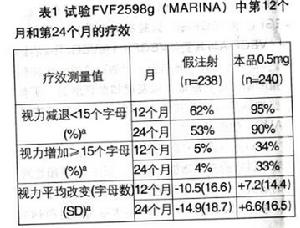
在新生血管性年齡相關性黃斑變性(AMD)患者中進行的三項隨機、雙盲、假注射*或陽性對照的研究,評估了本品治療的臨床安全性與有效性。總計1,323名病人(879名本品治療組和444名對照組)入組至這三個臨床Ⅲ期試驗中。在研究FVF2598g(MARINA)中,輕微典型性或隱匿型不含典型性CNV患者每月接受本品0.3mg或0.5mg或假注射的玻璃體內注射。總計716名患者入組此研究(假注射治療組238名;本品0.3mg組238名;本品0.5mg組240名)。獲得了24個月的研究數據。
在研究FVF2587g(ANCHOR),典型性為主型CNV病變的患者接受:1)每月玻璃體內0.3mg本品注射與假PDT治療;2)每月玻璃體內0.5mg本品注射與假PDT治療;或者3)假注射玻璃體內注射與活性的維替泊芬PDT治療。本品首次注射後,同時給予假注射*的或活性的維替泊芬PDT治療,之後如果螢光素血管造影顯示受試眼中血管滲漏持續存在或複方,每三個月進行一次治療。總計423名患者入組此研究(假注射治療組143名;)本品0.3mg組140名;本品0.5mg組140名)。獲得至24個月的研究數據。
在兩項研究中,最初的有效性觀察指標為維持視力的患者比例,定義為第12個月時與基線相比視力下降小於15個字母。幾乎所有(約95%)接受本品治療的患者在治療12個月後保持了視力。34%至40%接受本品治療的患者出現有臨床意義的視力改善,定義為在第12個月,視力增加15個字母或以上。病變大小對結果無顯著的影響。詳細結果列於下表。
 雷珠單抗注射液
雷珠單抗注射液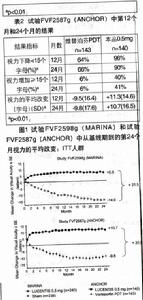 雷珠單抗注射液
雷珠單抗注射液在MARINA和ANCHOR研究中,第12個月時,0.5mg雷珠單抗治療組中觀察到視力改善的同時,亦伴隨了患者報告受益(以國立眼科研究所視功能調查問卷(VFQ-25)評分作為評價)。0.5mg雷珠單抗和兩個對照組之間的差異的p-值範圍為0.009至<0.0001。
兩項試驗的結果顯示,繼續雷珠單抗治療對於在治療第一年最佳矯正視力(BCVA)下降15個字母或以上的患者也有益。
對於使用本品治療超過36個月而言,尚未開展研究。
研究FVF3192g(PIER)為一項隨機、雙盲、假注射對照研究,目的是評價本品在所有類型的新生血管AMD患者中的安全性和有效性。患者在前三月內每月一次接受0.3mg(n=60)或0.5mg本品(n=61)玻璃體內注射或者玻璃體內假注射(n=63),之後每三月給藥一次。從第14個月開始,允許假注射的患者改為接受雷珠單抗治療,從第19個月開始,可能進行更頻繁的治療。在PIER研究中,本品治療的患者平均接受了10次治療。
主要療效終點為:相對於基線值,治療第12個月時視力的平均改變。在最初的視力增加之後(每月一次給藥後),在每三個月給藥一次期間患者視力普遍出現下降,在第12個月回到基線值,且在第24個月時大部分接受本品治療的患者(82%)保持這一療效。有些患者在一年假注射治療後改為接受雷珠單抗治療,其用藥情況表明,早期使用雷珠單抗或許能更好地維持視力。
在32位患者中開展了一項開放標籤研究(PROTECT),隨訪期為9個月,評價了維替泊芬PDT和0.5mg雷珠單抗同一天給藥的安全性。該研究的結果表明,初始治療後眼內炎症的發生率為6.3%(2/32)。
研究FVF3689g(SAILOR)為一項臨床Ⅲb期,單盲、一年期多中心研究,在之前未經治療和經治療的AMD繼發CNV受試者中進行。主要研究目標是評估接受12個月治療的受試者中,眼部和非眼部嚴重不良事件的發生率。2378名患者以1:1比例隨機分配接受每月一次玻璃體內0.3mg或0.5mg本品治療連續三個月,之後按需再治療,頻率不超過每月一次。
總體而言,兩個劑量組之間沒有觀察到眼部和非眼部不良事件的發生率有不平衡。統計學分析沒有顯示0.5mg組卒中發生率有增高的趨勢,各自的卒中總體發生率的95%置信區間較寬(0.3mg組為0.3%至1.3%,相比0.5mg組為0.7%至2.0%)。兩個給藥組中發生卒中的患者數量均較小,不能得出結論(或排除)治療組之間的卒中率確實有差異。卒中率的差異在具有已知卒中風險因子的患者,包括既往卒中病史或短暫性腦缺血發作史的患者中更大。
*假注射對照操作為使用與本品玻璃體內注射相同的方式進行麻醉,然後使用無針頭的注射器頂部壓向結膜,並推針。
藥理毒理
短尾猴雙側玻璃體腔內注射雷珠單抗0.25mg/眼至2.0mg/眼的劑量範圍,每2周一次,持續26周,出現劑量依賴的眼部影響。
注射後2天,前房閃輝與細胞出現劑量依賴性增加並達到峰值。炎性反應的程度在隨後注射治療中或恢復期內通常減輕,但並未在所有病例中完全恢復。在眼後節,發生玻璃體渾濁,該症狀同樣有劑量依賴性傾向,且一般持續至治療期結束。在26周的研究中,玻璃體炎症反應的嚴重程度隨注射次數而增加。恢復期後觀察到可逆的跡象。後節炎症的自然病程和持續時間顯示這是免疫介導的抗體反應,沒有臨床相關性。在相對長期的嚴重炎症後,在一些動物中觀察到白內障形成,提示晶狀體的改變更可能是繼發於嚴重的炎症。在玻璃體內注射藥物後觀察到眼內壓出現短暫的升高,這一反應與劑量大小無關。
眼組織的顯微鏡改變全部與炎症有關,沒有顯示有任何眼結構的變性過程。在有些眼的視神經盤中觀察到肉芽腫炎性改變。這些後段的變化在恢復期中減輕,有些病例中完全消失。本品玻璃體內注射後未發現全身毒性特徵。在部分接受給藥的動物中發現血清和玻璃體內中有本品的抗體。
尚無本品在動物中致癌、致突變和生殖毒性與發育毒性的數據。
藥代動力學
新生血管性AMD患者每月接受本品玻璃體內注射後,本品的血清濃度通常較低,血清濃度峰值(C)一般低於可50%抑制VEGF的濃度(11~27ng/ml,根據細胞增殖檢測的評估)。在0.05至1.0mg/眼的劑量範圍內血清Cmax與劑量成比例。
基於群體藥代動力學分析和本品在接受0.5mg劑量的患者的血清中的消除,本品在玻璃體力的平均消除半衰期約為9天。每月玻璃體內注射本品0.5mg/眼後,在給藥後約1天達血清C,預期一般範圍在0.79和2.90ng/ml之間,預期C一般範圍在0.07和0.49ng/ml之間。本品的血清濃度比玻璃體中的濃度低90000倍。
腎功能損傷患者:尚未在腎功能受損患者中進行本品藥代動力學的正式研究。在患者群體藥代動力學分析中,54%(389/725)為腎功能受損患者(39%為輕度,12%為中度,2%為重度)。在腎功能受損患者中,本品清除率的下降無臨床顯著意義。因此不需要進行劑量調整。
肝功能損傷:尚無有關本品在肝功能損害患者中藥代動力學的正式研究。
貯藏
2~8℃避光保存,不得冷凍。
請在兒童不可觸及的地方貯存。
包裝
1瓶/盒(10mg/ml,每瓶裝量為0.2ml);內附2個針頭和一支注射器(1個1ml無菌注射器,1個18G,5μm濾過針頭,用於抽取瓶內容物;1個30G注射針頭,用於進行玻璃體內注射)。
有效期
36個月。
執行標準
進口藥品註冊標準JS20100025
