概述
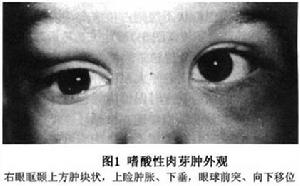 眼眶嗜酸性肉芽腫
眼眶嗜酸性肉芽腫眾多學者對該病進行了深入研究,從臨床表現和大量的超微結構和免疫組織化學的資料來看,現有傾向用朗格漢斯細胞組織細胞增多病(Langerhans cell histiocytosis)代替組織細胞增多病X。以前習慣性稱以上3種為不同類型的病變,現認為Letterer-Siwe病和Hand-Schüller-Christian病只是相同疾病的不同過程,嗜酸性細胞肉芽腫可能代表一種反應型,認為該組病變類似於肉芽腫的良性過程而不具有新生物的特性。根據預後好的特點,有理由將嗜酸性肉芽腫與其他兩種分離出來。現未發現孤立性的嗜酸性肉芽腫發展成為系統性多灶性病變或因此病而死亡的患者。病變沒有顯示家族性,屬於非遺傳性疾病急性或亞急性的嚴重病變可自發消退,小劑量的皮質類固醇、放射治療和抗腫瘤的化學治療能抑制病變的進程。根據臨床資料和病變的預後將該組織病變分為5類:
1.骨病變 50%的患者主要引起骨病變,單個或多個病灶傾向於累及軸性骨(頭顱脊柱、肩胛、骨盆和四肢近端長骨),多為嗜酸性細胞肉芽腫,預後良好。
2.骨病變和少量的全身病變 骨髓受累可導致貧血基底腦膜病變可引起尿崩症,黏膜和皮膚也受侵犯,此臨床表現在Hand-Schüller-Christian病中多見。
3.骨病變加上中到重度的內臟損害 骨損害主要發生在顱骨的基底部、眶周骨和中耳骨;內臟病變包括肺脾、肝以及淋巴結、黏膜和頭顱皮膚受累,該病變在嚴重的Hand-Schüller-Christian病和輕度的Leterer-Siwe病患者中發生。
4.嚴重的內臟病變 肝、脾臟迅速而進行性腫大,肺臟病、貧血、血小板減少和皮疹也很快出現,因為髓內組織細胞快速增生,故骨破壞比慢性病例發生少,這些發現與Letterer-Siwe病相符合預後差。
5.局限病變 這一小組患者一般很年輕,侵襲性病變發生在特殊的部位並伴有輕微的全身病變。這些病變可能是頸部的淋巴結病,肝臟病或少見的嚴重肺囊腫。
病因
發病機制不清,可能是一種免疫異常性疾病,特別與T淋巴細胞異常有關,單克隆抗體OKT-6可使Langerhans細胞表面膜受體染色。
臨床表現
孤立的骨病變多見於顱骨,依次為骨盆、脊柱、肋骨和四肢長骨。嗜酸性肉芽腫也可以是多灶性的。顱骨的額骨和頂骨是常見的病變區,當眶骨受累時外顳上眶緣是最普遍的部位,偶爾眶骨和顱骨廣泛受累病變區腫脹,有壓痛或疼痛,骨腫脹,骨右眼眶顳上方腫塊狀新生物,上瞼腫脹下垂。眼球前突、向下移位。病變發生在骨的板障層,突破眶骨膜,引起上瞼外側軟組織炎症和淚腺發炎,故臨床類似於眼眶的皮樣囊腫和淚腺炎的表現。極個別病變可累及角膜鞏膜和葡萄膜。
併發症:
肉芽腫侵及角膜、鞏膜或葡萄膜時可出現相應的不同症狀。
分類
眼眶嗜酸性肉芽腫是由大量增生的染色淡而大的組織細胞在眼眶或全身各臟器軟組織和骨中堆積引起的病變。根據累及的部位不同又分為局部單灶性和全身多灶性。年齡越小越容易發展成為多灶性病灶患者年齡偏大,單灶性病變可能性大。
嗜酸性細胞肉芽腫可能代表一種反應型,認為該組病變類似於肉芽腫的良性過程,而不具有新生物的特性。根據預後好的特點,有理由將嗜酸性肉芽腫與其他兩種分離出來。現未發現孤立性的嗜酸性肉芽腫發展成為系統性多灶性病變或因此病而死亡的患者。病變沒有顯示家族性,屬於非遺傳性疾病。急性或亞急性的嚴重病變可消退,小劑量的皮質類固醇、放射治療和抗腫瘤的化學治療能抑制病變的進程。根據臨床資料和病變的預後將該組織病變分為5類:
1.骨病變50%的患者主要引起骨病變,單個或多個病灶傾向於累及軸性骨(頭顱、脊柱、肩胛、骨盆和四肢近端長骨),多為嗜酸性細胞肉芽腫,預後良好。
2.骨病變和少量的全身病變骨髓受累可導致貧血,基底腦膜病變可引起,黏膜和皮膚也受侵犯,此臨床表現在Hand-Schüller-Christian病中多見。
3.骨病變加上中到重度的內臟損害骨損害主要發生在顱骨的基底部、眶周骨和中耳骨;內臟病變包括肺、脾、肝以及淋巴結、黏膜和頭顱皮膚受累,該病變在嚴重的Hand-Schüller-Christian病和輕度的Leterer-Siwe病患者中發生。
4.嚴重的內臟病變肝、脾臟迅速而進行性腫大,肺臟病、貧血、血小板減少和皮疹也很快出現,因為髓內組織細胞快速增生,故骨破壞比慢性病例發生少,這些發現與Letterer-Siwe病相符合,預後差。
5.局限病變這一小組患者一般很年輕,侵襲性病變發生在特殊的部位並伴有輕微的全身病變。這些病變可能是頸部的淋巴結病,肝臟病或少見的嚴重肺囊腫。
診斷
 眼眶嗜酸性肉芽腫
眼眶嗜酸性肉芽腫患者常為兒童,在顳上外眶緣處捫及腫塊,有壓痛,就應想到嗜酸性肉芽腫的可能性X線照片顯示不規則、鋸齒狀的溶骨區,無硬化邊界。CT檢查除發現溶骨性缺損外,還發現外上方軟組織密度增加,局部高起。對這樣的病變應做活體組織檢查,發現大量的組織細胞增生,電子顯微鏡見細胞質內有Langerhans顆粒,對年齡較大的患者可診斷嗜酸性肉芽腫。
鑑別診斷:
在組織病理學上有2個病變顯示組織細胞。一個是“膽固醇瘤”發生於眶周的骨內;另一個是巨細胞修復性肉芽腫膽固醇肉芽腫在組織病理學上不顯示嗜伊紅細胞可以與嗜伊紅細胞肉芽腫鑑別。巨細胞修復性肉芽腫則顯示區域性形狀,有出血、纖維化和多核巨細胞區,並且也無嗜伊紅細胞。
檢查
 眼眶嗜酸性肉芽腫
眼眶嗜酸性肉芽腫實驗室檢查:
1.免疫學檢查 可能發現部分指標異常。
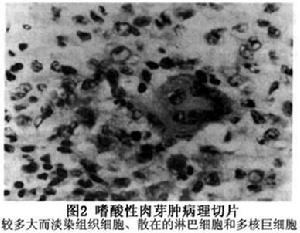
2.病理學檢查 活檢標本的組織病理學顯示最明顯的細胞是大而淡染的組織細胞,核位於細胞中心,呈卵圓形,空泡狀核膜輕度凹陷,嗜酸性粒細胞質內有較多的顆粒。在組織細胞浸潤的背景中,可見不同數量的嗜酸性粒細胞淋巴細胞、漿細胞和多核巨細胞。病灶區內基質少,但血管多,易出血,故可見含鐵血黃色素的巨噬細胞。舊病變中的單核細胞和多核細胞發生脂化,骨內的一些病變可以自愈,病變區纖維化而呈現硬化灶。
電子顯微鏡證實單核細胞組織細胞質內有特殊的顆粒呈棒狀,中心有條紋,末端有開口的空泡,酷似球拍故叫球拍小體又名Birbeck顆粒或Langerhans顆粒。多核巨細胞含有豐富的線粒體、滑面內質網,但無Langerhans顆粒。所以Langerhans顆粒是樹突狀組織細胞(Langerhans細胞)的標記。現已知嗜酸性細胞肉芽腫是Langerhans細胞的增生,該細胞常存在皮膚的表皮層,這就可解釋嗜酸性細胞肉芽腫的組織細胞高度嗜表皮的原因。
其它輔助檢查:
1.X線檢查 顱骨是最好發部位,常侵及多塊顱骨尤以顱蓋骨為甚。病灶大小不等,常相互融合呈大塊“地圖樣”骨缺損,這種表現頗有特徵。顱骨表現多從板障開始隨病灶的擴展,使內外骨板破壞,局部軟組織隆起腫脹,骨質破壞區的邊緣銳利,通常無硬化,也無骨膜反應是其特點。眶窩骨質破壞以累及眼眶之外上緣為多,並出現同側突眼。
2.超音波探查 病變累及眼眶時眼眶內可探及異常回聲區,邊緣不清,形態不規則,內回聲強弱分布不均勻,聲衰減明顯,後界顯示較弱或不清缺乏可壓縮性。
3.CT掃描 眶骨骨質破壞呈溶骨性,邊緣清晰,局部軟組織腫脹可以侵犯眼外肌、淚腺、甚至眼球,顳肌亦可侵犯,增強後CT掃描呈中度到明顯強化。骨破壞常發生在眶前或前顳部,顳鱗部及眶骨骨質破壞均可引起眼球突出。
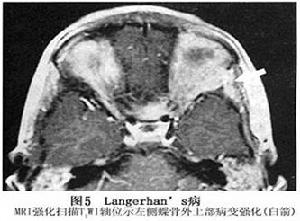
4.MRI 病變常位於肌錐外,表現為不規則腫塊,病變發展到一定程度可破壞眶壁,侵及顳窩或顱腔侵及顱內者,類似其他顱眶溝通瘤表現,但境界多較清楚,T1加權像呈中低信號,T2加權像呈中高信號。腫塊侵及眼外肌致其受壓移位,境界不清眼球常有突出。
治療
眼眶嗜酸性肉芽腫為單灶性病變,手術刮除能消除病灶,皮質類固醇內注射能治癒病灶,但注射時切勿穿過硬腦膜,將藥物注射到顱內。有作者主張在病灶刮除後,給局部小劑量分次放射治療,總劑量6~8Gy,效果更好也可以觀察,因病變為良性,且有自愈傾向。對復發病例要全身用皮質類固醇和抗代謝藥物治療,Song等報導1例9歲男孩眼眶外上方嗜酸性肉芽腫刮除術後6個月,MRI檢查發現眶內腫塊復發,用潑尼松和長春新鹼治療,效果顯著。
預後
本病的預後與發病時的年齡、病變程度是否伴有器官功能障礙等情況有關。發病年齡越早預後越差。本病的部分病例可以自行緩解。治療可根據病情輕重採用保守療法、激素療法和化學療法亦可考慮小劑量放射治療。
