
概述
幼年性黃色肉芽腫是一種良性、自限性黃棕色丘疹或結節。常於出生後或生後一年內出現,多發性,12~18個月內自然消退。少數可以發生於眼、肺、心包、腦膜、肝、脾等處。幼年性黃色肉芽腫(juvenilexanthogranuloma)也稱青少年黃色肉芽腫,以前稱痣黃內皮瘤(nevoxxanthoendothelioma)或先天性黃色瘤複合體(congenitalxanthomamultiplex),是一種良性組織細胞增多所引起的皮膚、眼部和眼眶病變,治療效果好,並有自愈傾向。
臨床表現
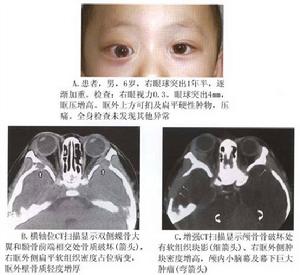 幼年性黃色肉芽腫
幼年性黃色肉芽腫本病發生於嬰兒和兒童,皮膚和眼是好發部位,很少侵犯內臟。頭、頸和四肢近端皮膚常受到病變的侵犯,皮膚病變常為圓形高起,橘黃色褐色或紅藍色結節。眼臉皮膚結節可能是孤立的也可能是面部皮膚結節的一部分,約1/4病人出現多個結節,一般不會超過10個,不會發生潰瘍。約1/5的患者在出生時就有皮膚結節,2~5歲時消失。病變可累及肺臟、心包縱隔、腹膜後組織、橫紋肌、胃腸和睪丸,但一般比較少見。
眼部表現:一般不易發生眼內病變。結膜、角膜和鞏膜等眼球表面的病變比眼瞼的病變少得多,眼球表面病變一般不與眼內的病變同時存在眼瞼病變的發病年齡較大,眼球表面和眼內病變同時發病者年齡偏小。在嬰兒中眼內病變常發生在虹膜和睫狀體,引起自發性前房積血,雖然病變可自發消退,對生命沒有影響,但反覆前房積血,可引起青光眼而造成視力喪失。虹膜病變可能是結節性或彌散性也可引起虹膜異色。脈絡膜和視網膜受累少見,85%的眼病患兒小於1歲眼病可能在皮膚病以前出現。
眼眶受侵犯少見,病變可侵犯眶骨和蝶骨,產生嚴重的骨質破壞病變可位於眶前部或眶後部,約50%的眶內病變侵犯眼外肌,視神經也可受到侵犯。約1/6的皮膚病變的患者患有眼眶病。幼年性黃色肉芽腫與神經纖維瘤病有某種關聯,在患者軀幹皮膚可發現牛奶咖啡斑,偶爾在眼眶發現神經纖維瘤。
雖然眼眶侵犯較少,但可侵及骨質,產生嚴重的骨破壞。眼部症狀包括眼球突出、眶周可觸及腫塊等眼眶占位病變症狀和體徵。病變也可累及顱內顯示多發灶由於病灶較小,多無顱內症狀。
併發症:
肉芽腫病變可累及內臟如胃腸等組織,出現相應的臨床表現。眼部反覆出血可導致繼發性青光眼。
診斷
嬰兒和兒童面部和頸部皮膚有高起橘黃色或紫紅色的結節,前房有自發性出血或眼眶受累,這些是幼年性黃色肉芽腫的典型臨床表現。最好能切除皮膚結節進行組織病理學檢查,病變內能發現大量的組織細胞和散在的Touton多核巨細胞便可確立診斷。
鑑別診斷:
但應與組織細胞增多症X相區別,患者內臟系統很少受累與Letterer-Siwe病不同;無地圖樣顱骨破壞和尿崩症可與Hand-Schüller-Christian病相鑑別;組織細胞質內無Langerhans顆粒可和嗜酸性細胞肉芽腫相區別;患者頸部淋巴結不腫大,與竇性組織增多病不一樣。
檢查
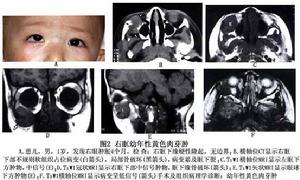 幼年性黃色肉芽腫
幼年性黃色肉芽腫實驗室檢查:
1.血液常規檢查 了解血細胞數量,必要時行骨髓穿刺,根據其結果對鑒診斷有用。
2.病理組織學檢查 發現病變內主要是比較正常的組織細胞細胞較大,核呈卵圓形,細胞質染色淡,部分細胞質脂化呈泡沫狀,也有部分組織細胞呈梭形。在組織細胞的背景中可見散在的淋巴細胞、漿細胞和少許的嗜酸性細胞。該病顯著的組織病理學特徵是Touton多核巨細胞,多核呈環狀位於細胞的中心,核環中心區有均質的嗜酸性胞質,核環周圍的細胞質可呈泡沫狀,這些Touton多核巨細胞多見於皮膚和眼眶組織的病變中皮膚和眼眶病變組織中血管少,故出血機會不多。虹膜病變內的血管多,且位於病變的表面,可被誤認為虹膜血管瘤,血管壁薄所以容易引起前房積血。電子顯微鏡檢查在組織細胞質內未見到Langerhan顆粒陳舊性病變以大量成纖維細胞增生形成瘢痕。
其它輔助檢查:
影像學檢查:超聲、CT和MRI很類似朗格漢斯細胞組織細胞增生症,表現為眼眶軟組織塊影,眶骨及顱內多發性骨破壞。MRI顯示眶顱多發病灶如骨破壞明顯,X線可顯示顱內多發性骨破壞病灶,邊界清楚,但不整齊。
治療
該病對治療反應好,局部病變可手術切除,眼眶病變採用小劑量的放射或糖皮質激素局部或全身治療。前房大量出血、眼壓高,可引起角膜血染和視力喪失的病例除用降眼壓藥物外,適當時可前房穿刺行血液或血塊取出。
如病變非常廣泛,特別是皮膚大量結節出現,而眼部病變對眼功能無影響,治療有困難或治療有明顯副作用時,可隨訪觀察,因幼年性黃色肉芽腫是非侵襲性病變,並可自發好轉皮膚病變4~6歲時逐漸消失青春期時所有病變完全自愈。
