
方法定義
 氣相色譜法
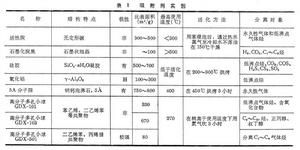
氣相色譜法氣相色譜法由於所用的固定相不同,可以分為兩種,用固體吸附劑作固定相的叫氣固色譜,用塗有固定液的單體作固定相的叫氣液色譜。
按色譜分離原理來分,氣相色譜法亦可分為吸附色譜和分配色譜兩類,在氣固色譜中,固定相為吸附劑,氣固色譜屬於吸附色譜,氣液色譜屬於分配色譜。
按色譜操作形式來分,氣相色譜屬於柱色譜,根據所使用的色譜柱粗細不同,可分為一般填充柱和毛細管柱兩類。一般填充柱是將固定相裝在一根玻璃或金屬的管中,管內徑為2~6毫米。毛細管柱則又可分為空心毛細管柱和填充毛細管柱兩種。空心毛細管柱是將固定液直接塗在內徑只有0.1~0.5毫米的玻璃或金屬毛細管的內壁上,填充毛細管柱近幾年才發展起來的,它是將某些多孔性固體顆粒裝入厚壁玻管中,然後加熱拉製成毛細管,一般內徑為0.25~0.5毫米。
在實際工作中,氣相色譜法是以氣液色譜為主。
方法正文
 氣相色譜法
氣相色譜法分析法概述
氣相色譜儀是用於分離複雜樣品中的化合物的化學分析儀器。氣相色譜儀中有一根流通型的狹長管道,這就是色譜柱。在色譜柱中,不同的樣品因為具有不同的物理和化學性質,與特定的柱填充物(固定相)有著不同的相互作用而被氣流(載氣,流動相)以不同的速率帶動。當化合物從柱的末端流出時,它們被檢測器檢測到,產生相應的信號,並被轉化為電信號輸出。在色譜柱中固定相的作用是分離不同的組分,使得不同的組分在不同的時間(保留時間)從柱的末端流出。其它影響物質流出柱的順序及保留時間的因素包括載氣的流速,溫度等。在氣相色譜分析法中,一定量(已知量)的氣體或液體分析物被注入到柱一端的進樣口中(通常使用微量進樣器,也可以使用固相微萃取纖維(solidphasemicroextractionfibres)或氣源切換裝置)。當分析物在載氣帶動下通過色譜柱時,分析物的分子會受到柱壁或柱中填料的吸附,使通過柱的速度降低。分子通過色譜柱的速率取決於吸附的強度,它由被分析物分子的種類與固定相的類型決定。由於每一種類型的分子都有自己的通過速率,分析物中的各種不同組分就會在不同的時間(保留時間)到達柱的末端,從而得到分離。檢測器用於檢測柱的流出流,從而確定每一個組分到達色譜柱末端的時間以及每一個組分的含量。通常來說,人們通過物質流出柱(被洗脫)的順序和它們在柱中的保留時間來表征不同的物質。檢測器
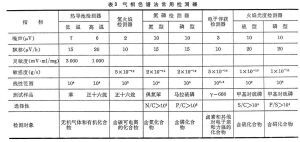
氣相色譜法中可以使用的檢測器有很多種,最常用的有火焰電離檢測器(FID)與熱導檢測器(TCD)。這兩種檢測器都對很多種分析成分有靈敏的回響,同時可以測定一個很大的範圍內的濃度。TCD從本質上來說是通用性的,可以用於檢測除了載氣之外的任何物質(只要它們的熱導性能在檢測器檢測的溫度下與載氣不同),而FID則主要對烴類回響靈敏。FID對烴類的檢測比TCD更靈敏,但卻不能用來檢測水。兩種檢測器都很強大。由於TCD的檢測是非破壞性的,它可以與破壞性的FID串聯使用(連線在FID之前),從而對同一分析物給出兩個相互補充的分析信息。其它的檢測器要么只能檢測出個別的被測物,要么可以測定的濃度範圍很窄。常見的檢測器包括:
放電離子化檢測器(DID),它通過高壓放電來產生離子。
電子俘獲檢測器,它使用β放射線源(電子流)來測量樣品對電子的俘獲能力。
火焰光度檢測器(FPD)
火焰電離檢測器(FID)
霍爾電導檢測器(ElCD)
氦離子化檢測器(HID)
氮磷檢測器(NPD)
質譜檢測器(MSD)
光離子化檢測器(PID)
脈衝放電檢測器(PDD)
熱能(熱導)分析器/檢測器(TEA/TCD)有一些氣相色譜儀與質譜儀相連線而以質譜儀作為它的檢測器,這種組合的儀器稱為氣相色譜-質譜聯用(GC-MS,簡稱氣質聯用),有一些氣質聯用儀還與核磁共振波譜儀相連線,後者作為輔助的檢測器,這種儀器稱為氣相色譜-質譜-核磁共振聯用(GC-MS-NMR)。有一些GC-MS-NMR儀器還與紅外光譜儀相連線,後者作為輔助的檢測器,這種組合叫做氣相色譜-質譜-核磁共振-紅外聯用(GC-MS-NMR-IR)。但是必須指出,這種情況是很少見的,大部分的分析物用單純的氣質聯用儀就可以解決問題。
通俗色譜
電影,書籍與電視節目經常歪曲氣相色譜法的能力以及運用氣相色譜法完成的工作。例如,在美國的電視節目《鑑證行動組》中,人們用氣相色譜來快速地識別未知樣品。分析員在取得樣品之後十五分鐘之後就會說:“這是在過去兩個星期中在雪佛龍公司(Chevron)的油站里購買的汽油。”事實上,一個典型的氣相色譜分析所用的時間要長得多。有時依照選定的程式,一個樣品就要進行一個多小時的分析。對色譜柱進行“清理”以便接受下一個樣品還需要額外的時間。同時,為了驗證一個結論,分析員往往需要進行多次平行的分析,因為單次分析的結果很可能具有偶然性(參見顯著性差異)。同時,氣相色譜並不能識別大部分的樣品,而且並非樣品中的所有物質都可以通過氣相色譜檢測出來。氣相色譜真正能告訴分析者的,只是在某個時間有一種物質從色譜柱中被洗脫出來,而且檢測器對它有回響。為了使結果變得更有意義,分析人員需要知道樣品中可能含有什麼成分,以及它們可能有怎么樣的濃度。還有,一些低含量的物質可能因為與另一種高含量的物質同時被洗脫而無法在色譜圖中表現出來。最後,分析人員還經常需要將未知樣品的氣相色譜結果與可能存在的物質的標準樣品的分析結果進行比較。氣相色譜-質譜聯用儀可以很好地改善這種混淆不清的狀況,因為質譜儀可以識別出各組分的相對分子質量。不過,要很好地完成這些工作,同樣需要時間與技巧。類似地,絕大部分的氣相色譜分析並不是簡單的按鍵操作。你不能簡單地將樣品瓶放在自動採樣器的托盤上,然後按一個按鈕,讓計算機告訴你關於樣品的所有信息。根據被分析的物質,分析人員需要小心選擇一套合適的操作程式。不過也要承認,在對相似樣品的大量重複性分析之中,簡單的按鍵操作是存在的。這包括化工生產中的分析,也包括為了確定樣品中被測物的平均含量而對同一實驗獲得的20個樣品的分析等等。不過,那些書籍,電影與電視節目中的研究性工作絕對不屬於這種情況。方法原理
 氣相色譜法
氣相色譜法儀器裝置
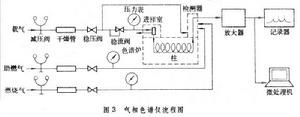
氣流系統
 氣相色譜儀流程圖
氣相色譜儀流程圖分離系統
 氣相色譜法
氣相色譜法檢測系統
包括檢測器、微電流放大器、記錄器。檢測器(表3)將色譜柱流出的組分,依濃度的變化轉化為電信號,經微電流放大器後,把放大後的電信號分別送到記錄器和數據處理裝置,由記錄器繪出色譜流出曲線。數據處理系統
簡單的數據處理部件是積分儀。新型的氣相色譜儀都有微處理機作數據處理。溫度控制系統及其他輔助部件溫度控制器用於控制進樣室、色譜柱、檢測器的溫度。如果色譜柱放置在有鼓風的色譜爐內,則要求色譜爐能在恆定溫度或程式升溫下操作。重要的輔助部件有頂空取樣器、流程切換裝置等。流動相
即載氣,可用氦氣、二氧化碳、氫氣、氮氣等。載氣的選擇與純化的要求取決於所用的色譜柱、檢測器和分析項目的要求,如對有些固定相不能與微量氧氣接觸,又如對熱傳導池檢測器宜用氫氣作載氣;對電子捕獲檢測器須除去載氣中負電性較強的雜質,以利於提高檢測器的靈敏度。用分子量小的氣體作載氣時可用較高的線速,這時柱效下降不大,卻可以縮短分析時間,因為分子量小的氣體粘度小,柱壓增加不大,並且在高線速時可減小氣相傳質阻力。用氫氣作載氣時,在填充柱和開管柱中的流速可分別選用35和2毫升/分左右。固定相
一般來說,宜按“相似性”原則選擇固定液;分析非極性樣品時用非極性固定液;分析強極性樣品時用極性強的固定液(表4)。把固定液塗敷於開管柱的內壁,或塗漬在載體上製成填充柱的固定相,均勿太厚。開管柱的df宜為0.2~0.4微米,填充柱的固定液含量宜為3%~10%。載體顆粒約為柱徑的0.1,即80~100目較好。這樣,組分在液相中傳質快,載體粒度較小而又未增大填充不均勻性,有利於在較低的溫度下分析高沸點組分及縮短分析時間。操作溫度
進樣室的溫度應根據進樣方法和樣品而定。氣化方式進樣時,氣化溫度既要使組分能充分氣化,又不會分解(裂解進樣除外)。檢測室的溫度以稍高於柱溫為好,可避免組分冷凝或產生其他問題。色譜柱溫的確定要作綜合考慮,即要照顧到固定相的使用溫度範圍、分析時間長短、便於定性和定量測定等因素。最好能在恆溫下操作,沸程很寬的樣品才採用程式升溫操作。滿意的操作溫度須由實驗求得。樣品預處理
欲分析的化合物常用化學反應的方法轉變成另一種化合物,這稱為衍生物的製備。然後再對衍生物進行色譜分析。預處理的好處是:①許多化合物揮發性過低或過高,極性很小或熱穩定性差,不能或不適於直接取樣注入色譜分析儀進行分析,其衍生物則可以很方便地進入色譜儀;②一些難於分離的組分,轉化成衍生物就便於分離和進行定性分析;③用選擇性檢測器檢測可獲得高靈敏度的衍生物;④樣品中有些雜質因不能成為衍生物而被除去。氣相色譜法最常用的化學衍生物法有矽烷化反應法、醯化反應法和酯化反應法(有重氮甲烷法、三氟化硼催化法和季硼鹽分解法等)。在製備化學衍生物時要特別仔細,否則會帶來嚴重的錯誤。
色譜分析
綜述
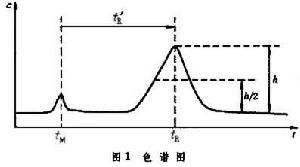
從色譜圖可以看到,色譜峰是組分在色譜柱運行的結果,它是判斷組分是什麼物質及其含量的依據,色譜法就是依據色譜峰的移動速度和大小來取得組分的定性和定量分析結果的。
定性分析
在給定的條件下,表示組分在色譜柱內移動速度的調整保留時間是判斷組分是什麼物質的指標,即某組分在給定條件下的t惱值必定是某一數值(圖1)。為了儘量免除載氣流速、柱長、固定液用量等操作條件的改變對使用t惱值作定性分析指標時產生的不方便,可進一步用組分相對保留值α或組分的保留指數來進行定性分析。計算組分i在給定的柱溫和固定相時的保留指數Ii的公式為。式中n與n+1是緊靠在組分i前後流出的正構烷烴的碳原子數,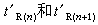 是這兩個正構烷烴的調整保留時間。將樣品進行色譜分析後,按同樣的實驗條件用純物質作實驗,或者查閱文獻,把兩者所得的定性指標(α值、t惱值或I值)相比較,如果樣品和純物質都有定性指標數值一致的色譜峰,則此樣品中有此物質。由於只能說相同物質具有相同保留值的色譜峰,而不能說相同保留值的色譜峰都是一種物質,所以為了更好地對色譜峰進行定性分析,還常採用其他手段來直接定性,例如採用氣相色譜和質譜或光譜聯用,使用選擇性的色譜檢測器,用化學試劑檢測和利用化學反應等。
是這兩個正構烷烴的調整保留時間。將樣品進行色譜分析後,按同樣的實驗條件用純物質作實驗,或者查閱文獻,把兩者所得的定性指標(α值、t惱值或I值)相比較,如果樣品和純物質都有定性指標數值一致的色譜峰,則此樣品中有此物質。由於只能說相同物質具有相同保留值的色譜峰,而不能說相同保留值的色譜峰都是一種物質,所以為了更好地對色譜峰進行定性分析,還常採用其他手段來直接定性,例如採用氣相色譜和質譜或光譜聯用,使用選擇性的色譜檢測器,用化學試劑檢測和利用化學反應等。定量分析
色譜峰的大小由峰的高度或峰的面積確定。可用手工的方法測量峰高,和以峰高h與峰高一半處的峰寬ω┩的乘積表示峰面積。A=hω┩。新型的色譜儀都有積分儀或微處理機給出更精確的色譜峰高或面積。應該注意,組分進入檢測器產生的相應的色譜信號大小(峰高或峰面積)隨所用檢測器類別和載氣的不同而異,有時甚至受到物質濃度和儀器結構的影響。所以須將所得的色譜信號予以校正,才能與組分的量一致,即需要用下式校正組分的重量:W=f′A式中f′為該組分的定量校正因子。依上式從色譜峰面積(或峰高)可得到相應組分的重量,進一步用下述方法之一計算出組分i在樣品中的含量Wi:①歸一化法,將組分的色譜峰面積乘以各自的定量校正因子。此法的優點是方法簡便,進樣量與載氣流速的影響不大;缺點是樣品中的組分必須在色譜圖中都能給出各自的峰面積,還必須知道各組分的校正因子。②內標法,向樣品中加入被稱為內標物的某物質後,進行色譜分析,然後用它對組分進行定量分析。例如稱取樣品Wm克,將內標物Wφ克加入其中,進行色譜分析後,得到欲測定的組分與內標物的色譜峰面積分別為Ai和Aφ。此方法沒有歸一化法的缺點,不足之處是要求準確稱取樣品和內標物的重量,選擇合適的內標物。
③外標法,在進樣量、色譜儀器和操作等分析條件嚴格固定不變的情況下,先用組分含量不同的純樣等量進樣,進行色譜分析,求得含量與色譜峰面積的關係,用下式進行計算:式中k媴是組分i單位峰面積百分含量校正值。此法適用於工廠控制分析,特別是氣體分析;缺點是難以做到進樣量固定和操作條件穩定。
優點缺點
優點
①分離效率高,分析速度快,例如可將汽油樣品在兩小時內分離出200多個色譜峰,一般的樣品分析可在20分種內完成。②樣品用量少和檢測靈敏度高,例如氣體樣品用量為1毫升,液體樣品用量為0.1微升,固體樣品用量為幾微克。用適當的檢測器能檢測出含量在百萬分之十幾至十億分之幾的雜質。③選擇性好,可分離、分析恆沸混合物,沸點相近的物質,某些同位素,順式與反式異構體,鄰、間、對位異構體,鏇光異構體等。④套用範圍廣,雖然主要用於分析各種氣體和易揮發的有機物質,但在一定的條件下,也可以分析高沸點物質和固體樣品。套用的主要領域有石油工業、環境保護、臨床化學、藥物學、食品工業等。缺點
為在對組分直接進行定性分析時,必須用已知物或已知數據與相應的色譜峰進行對比,或與其他方法(如質譜、光譜)聯用,才能獲得直接肯定的結果。在定量分析時,常需要用已知物純樣品對檢測後輸出的信號進行校正。方法展望
今後氣相色譜法還將有很大的發展,耐高溫的極性高效開管柱和選擇性好、靈敏度高的檢測器的研製,色譜定性和定量分析規律的研究,微處理機進一步的套用,生物學、醫學、環境保護等方面新的分析方法都是很活躍的研究課題。智慧型氣相色譜法的研究也是今後發展的方向。儀器要求
所用的儀器為氣相色譜儀。除另有規定外,載氣為氮氣;色譜柱為填充柱或毛細管柱,填充柱的材質為不鏽鋼或玻璃,載體用直徑為0.25~0.18mm、0.18~0.15mm或0.15~0.125mm經酸洗並矽烷化處理的硅藻土或高分子多孔小球;常用玻璃或彈性石英毛細管柱的內徑為0.20或0.32mm。進樣口溫度應高於柱溫30~50℃;進樣量一般不超過數微升;柱徑越細進樣量應越少。檢測器為氫火焰離子化檢測器,檢測溫度一般高於柱溫,並不得低於100℃,以免水氣凝結,通常為250~350℃。正文中各品種項下規定的條件,除檢測器種類、固定液品種及特殊指定的色譜柱材料不得任意改變外,其餘如色譜柱內徑、長度、載體牌號、粒度、固定液塗布濃度、載氣流速、柱溫、進樣量、檢測器的靈敏度等,均可適當改變,以適應具體品種並符合系統適用性試驗的要求。一般色譜圖約於30分鐘內記錄完畢。方法分類
內標準法
取標準被測成分,按依次增加或減少的已知階段量,各自分別加入各單體所規定的定量內標準物質中,調製標準溶液。分別取此標準液的一定量注入色譜柱,根據色譜圖取標準被測成分的峰面積和峰高和內標物質的峰面積和峰高的比例為縱坐標,取標準被測成分量和內標物質量之比,或標準被測成分量為橫坐標,製成標準曲線。然後按單體中所規定的方法調製試樣液。在調製試樣液時,預先加入與調製標準液時等量的內標物質。然後按製作標準曲線時的同樣條件下得出的色譜,求出被測成分的峰面積或峰高和內標物質的峰積或峰高之比,再按標準曲線求出被測成分的含量。所用的內標物質,應採用其峰面積的位置與被測成分的峰的位置儘可能接近並與被測成分以外的峰位置完全分離的穩定的物質。絕對標準曲線法
取標準被測成分按依次增加或減少階段法,各自調製成標準液,注入一定量後,按色譜圖取標準被測成分的峰面積或峰高為縱坐標,而以標準被測成分的含量為橫坐標,製成標準曲線。然後按單體中所規定的方法製備試樣液。取試樣液按制標準曲線時相同的條件作出色譜,求出被測成分的峰面積和峰高,再按標準曲線求出被測成分的含量。峰面積百分率法
以色譜中所得各種成分的峰面積的總和為100,按各成分的峰面積總和之比,求出各成分的組成比率。分析方法
分析方法實際上是在某一特定的氣相色譜分析中使用的一系列條件。建立分析方法實際上是確定對於某一分析的最佳條件的過程。為了滿足某一特定的分析的要求,可以改變的條件包括進樣口溫度,檢測器溫度,色譜柱溫度及其控溫程式,載氣種類及載氣流速,固定相,柱徑,柱長,進樣口類型及進樣口流速,樣品量,進樣方式等。檢測器還可能有其它可供調節的參數,這取決於所使用的檢測器類型。有一些氣相色譜儀還有可以控制樣品與載氣流向的閥門,這些閥門開啟與關閉的時間也可能對分析的效果有重要影響。右圖為GeoStrataTechnologies生產的Eclipse氣相色譜儀。它以三分鐘為周期持續運轉。該儀器有兩個閥門,用來控制載氣進入定量管。當定量管充滿樣品氣後,切換閥門,載氣就會通過定量管。載氣的壓強會將樣品帶入到色譜柱中進行分離。載氣選擇與載氣流速
典型的載氣包括氦氣、氮氣、氬氣、氫氣和空氣。通常,選用何種載氣取決於檢測器的類型。例如,放電離子化檢測器(DID)需要氦氣作為載氣。不過,當對氣體樣品進行分析的時候,載氣有時是根據樣品的母體選擇的,例如,當對氬氣中的混合物進行分析時,最好用氬氣作載氣,因為這樣做可以避免色譜圖中出現氬的峰。安全性與可獲得性也會影響載氣的選擇,比如說,氫氣可燃,而高純度的氦氣某些地區難以獲得。(參見:氦氣——分布與生產)很多時候,檢測器不僅僅決定了載氣的種類,還決定了載氣的純度(雖然對靈敏度的要求也在很大程度上影響載氣純度的要求)。通常來說,氣相色譜中所用的載氣,純度應該在99.995%以上。用於標識純度的典型商品名包括“零點氣級”,“高純度(UHP)級”,“4.5級”和“5.0級”。載氣流速對分析的影響在方式上與溫度類似(見下文)。載氣流速越高,分析速度越快,但是分離度越差。因此,最佳載氣流速的選擇與柱溫的選擇一樣,都需要在分析速度與分離度之間取得平衡。二十世紀九十年代之前生產的氣相色譜儀的載氣流速往往通過載氣入口的壓力(柱前壓)進行控制,實際的載氣流速則在柱的出口端通過電子流量計或皂膜流量計進行測定。這樣的一個過程常常很複雜,很耗時間,而且往往令人沮喪。在整個運行過程中,柱前壓不能再改變,氣流必須穩定。氣體流速與柱前壓的關係可以通過可壓縮流體的Poiseuille方程來計算。不過,很多現代的氣相色譜儀已經能用電路自動測定氣體流速,並通過自動控制柱前壓來控制流速。因此,載氣壓強與流速可以在運行過程中調整。柱前壓/氣流控制程式(與溫度控制程式類似)隨之出現。進樣口類型與流速
進樣口類型和進樣技術通常與樣品存在的形態(液態、氣態、被吸附、固態)以及是否存在需要氣化的溶劑有關。如果樣品分散良好,並且性質已知,那么它就可以通過冷柱頭進樣口直接進樣;如果需要蒸發除去部分溶劑,就使用分流/不分流進樣口(通常用注射器進樣);氣體樣品(如來自氣缸)通常用氣體閥進樣器進樣。被吸附的樣品(如在吸附管上)可以通過外部的(線上或離線)解吸裝置(如捕集-吹掃系統)或者在分流/不分流進樣器中解吸(使用固相微萃取技術)。樣品量與進樣技術
進樣技術氣相色譜中的十分之一原則真正的氣相色譜分析過程從樣品進入色譜柱開始。毛細管氣相色譜法的發展使得進樣技術面臨著很多實踐中的問題。柱上進樣技術多用於填充柱而不適用於毛細管柱。在毛細管氣相色譜儀中的進樣技術應該滿足以下兩個條件:進樣量不得超過柱的容量;與展開過程引起的樣品展寬相比,進樣後的塞式流寬度應該很小。如果不能滿足這一要求,色譜柱的分離能力將會下降。一個普遍的規則是,注入的體積,Vinj,和檢測器的體積,Vdet,應該只有樣品中包含被分析物的部分出柱時的體積的十分之一。以下是一些優秀進樣技術應當滿足的一般要求:應該能使色譜柱達到它的最佳分離效率;對於小量的有代表性的(典型)樣品,進樣應具有準確性和可重現性;不能改變樣品組成(對於具有不同的沸點、極性、濃度與熱力學穩定性的物質,進樣過程中不應有所差異);應該既適用於痕量分析,也適用於濃度相對較大的樣品。色譜柱的選擇
柱溫與溫度控制程式一個已經拆開以顯示出內部毛細管柱的氣相色譜儀恆溫箱氣相色譜儀中的色譜柱放置於溫度由電子電路精確控制的恆溫箱內。(當分析者說“柱溫”時,他實際上指的是恆溫箱的溫度。不過這種區別並不重要,因此在下文中對這兩者並不作區分。)樣品通過色譜柱的速率與溫度正相關。柱溫越高,樣品越快通過色譜柱。但是,樣品越快通過色譜柱,它與固定相之間的相互作用就越少,因此分離效果越差。通常來說,柱溫的選擇是綜合考慮分離時間與分離度的結果。柱溫在整個分析過程中不變的方法稱為恆溫方法。不過,在大部分的分析方法中,柱溫隨著分析過程的進行逐漸上升。初溫,升溫速率(溫度“斜率”)與末溫統稱為控溫程式。控溫程式使得較早被洗脫的被分析物能夠得到充分的分離,同時又縮短了較晚被洗脫的被分析物通過色譜柱的時間。
方法套用
只要在氣相色譜儀允許的條件下可以氣化而不分解的物質,都可以用氣相色譜法測定。對部分熱不穩定物質,或難以氣化的物質,通過化學衍生化的方法,仍可用氣相色譜法分析。在石油化工、醫藥衛生、環境監測、生物化學等領域都得到了廣泛的套用1.在衛生檢驗中的套用空氣、水中污染物如揮發性有機物、多環芳烴[苯、甲苯、苯並(a)比等];農作物中殘留有機氯、有機磷農藥等;食品添加劑苯甲酸等;體液和組織等生物材料的分析如胺基酸、脂肪酸、維生素等。2.在醫學檢驗中的套用體液和組織等生物材料的分析:如脂肪酸、甘油三酯、維生素、糖類等。3.在藥物分析中的套用抗癲癇藥、中成藥中揮發性成分、生物鹼類藥品的測定等。相關書籍
基本信息作者:李浩春、盧佩章定價:¥25.00元
出版社:科學出版社出版日期:1998年08月
ISBN:7-03-003123-7/O·572開本:32開
類別:分析化學及儀器頁數:391頁
簡介
本書共三篇十五章:第一篇十一章,敘述氣相色譜法的基本原理和技術,並對超臨界流體色譜法作了必要的介紹;第二篇兩章,介紹氣相色譜法柱系統的選擇方法,同時給出分析各類樣品的最佳色譜圖,以便進行解釋;第三篇兩章,論述氣相色譜法專家系統的目標、建立及開發情況.此外,還有8個附錄,詳細介紹氣相色譜法的各種固定相及各類化合物的保留指數.
目錄
第一篇氣相色譜法基礎
第一章緒論
1.1氣相色譜法的特點
1.2氣相色譜法術語
參考文獻
第二章氣相色譜儀
2.1填充柱氣相色譜儀
2.2毛細管柱氣相色譜儀
2.3製備型氣相色譜儀
2.4氣相色譜-質譜聯用儀(GC/MS)
2.5氣相色譜-傅立葉變換紅外光譜聯用儀(GC/FT-TR)
參考文獻
第三章氣相色譜氣流系統
3.1氣體
3.2氣體流量的控制
3.3氣體的純化
3.4氣體壓力與流量的測量
3.5載氣流量的校正
第四章氣相色譜分離系統
4.1固定相
4.1.1固體固定相
4.1.2液體固定相
4.2色譜柱
4.2.1填充色譜柱
4.2.2空心柱
4.3填充柱與空心柱的比較
參考文獻
第五章氣相色譜檢測系統
5.1色譜的檢測
5.2檢測器的性能與分類
5.2.1檢測器的性能要求
5.2.2檢測器的分類
5.3檢測器的評價
5.3.1噪聲與飄移
5.3.2檢測器的線性與線性範圍
5.3.3檢測器的靈敏度
5.3.4檢測呂的檢測限
5.3.5檢測器的最小檢測量和最小檢測濃度
5.3.6檢測器的回響時間
5.3.7檢測的相對靈敏度
5.4氣要色譜常用檢測器
5.4.1熱導檢測器(TCD)
5.4.2氫火焰離子化檢測器(FID)
5.4.3氮磷檢測器(NPD)
5.4.4電子俘獲檢測器(ECD)
5.4.5火焰光度檢測器(FPD)
參考文獻
第六章氣相色譜數據處理系統
6.1數據處理的目的和方法
6.2數據處理裝置
6.2.1記錄器
6.2.2積分儀
6.2.3數據處理站
6.3常用色譜軟體
參考文獻
第七章定性分析
7.1恆溫時色譜定性指標
7.1.1調整保留時間
7.1.2保留體積
7.1.3相對保留值
7.1.4保留指數
7.2程式升溫時色譜定性指標
7.3保留值定性法
7.3.1保留值對比定性法
7.3.2已知組分定性法
7.4檢測器定性法
7.4.1GC/MS定性
7.4.2GC/FT-IR定性
7.4.3選擇性檢測器定性
7.5化學試齊定性法
7.5.1消去法
7.5.2官能團法
7.6反應色譜定性法
7.6.1次甲基插入反應法
7.6.2氫化反應法
7.7保留值數據
7.7.1純樣品測定
7.7.2保留值規律
參考文獻
第八章定量分析
8.1色譜峰的測量
8.2定量校正因子
8.2.1分類與換算
8.2.2校正因子的測定
8.2.3面積相對校正因子的計算方法
8.3定量計算方法
8.3.1歸一化法
8.3.2內標法
8.3.3外標法
8.4影響準確定量的主要因素
參考文獻
第九章輔助技術
9.1柱切換技術
9.1.1坪面閥住切換技術
9.1.2壓力管柱切換技術
9.2化學衍生方法
9.2.1矽烷化反應法
9.2.2醯化反應法
9.2.3酯化反應法
9.3進樣技術
9.3.1分流式進樣法
9.3.2Grob式不分流進樣法
9.3.3直接進樣法
9.3.4冷卻式柱上進樣法
9.4頂空分析法
9.5裂解色譜法
參考文獻
第十章超臨界流體色譜法
10.1特點
10.2裝置
10.3操作條件
10.4套用
參考文獻
第十一章儀器的調試與維修
11.1儀器的調式
11.1.1準備工作
11.1.2氣路的考查
11.1.3電器件的考查
11.1.4檢測器的測試
11.1.5定性、定量測定
11.1.6柱效能與拖尾因子測定
11.2儀器的維修
參考文獻
第二篇氣相色譜法柱系統
第十二章柱系統的選擇
12.1柱效與操作條件
12.2固定相與樣品組分
參考文獻
第十三章色譜分析的套用
13.1碳氫化合物
13.2有機含氧化合物
13.3有機含氮化合物
13.4有機含硫化合物
13.5含鹵素化合物
13.6元素有機化合物
13.7農藥
13.8高分子材料
13.9藥物
13.10香料和精油
13.11臨床醫學樣品
13.12食品
13.13環境保護
13.14無機氣體
13.15同位素
參考文獻
第三篇氣相色譜專家系統
第十四章色譜專家系統的目標
14.1問題的提出
14.2問題的解決
第十五章色譜專家系統的建立
15.1專家系統的定義
15.2專家系統的結構
15.3專家系統的開發過程
15.4專家系統的語言
15.5專家系統開發工具
15.6氣相色譜專家系統
參考文獻
附錄
附錄1氣相色譜法常用符號
附錄2水蒸氣飽和蒸汽壓
附錄3壓力梯度校正因子(j)
附錄4常用高分子多孔小球固定相
附錄5常用化學鍵合固定相
附錄6氣相色譜法重要載體
附錄7氣相色譜法重要固定液
附錄8各類化合物保留指數索引
