流行病學
本病多在6~8歲發病。目前沒有其他相關內容描述。病因
本病的病因是常染色體隱性遺傳。發病機制
 黏多糖貯積症Ⅴ型
黏多糖貯積症Ⅴ型病理:本型的病理改變在某些方面與MPSⅠ型相似顯微鏡檢查肝組織肝實質細胞和庫普弗細胞含有大量空泡。脾臟結締組織增加,脾竇周圍脂肪軟骨細胞營養不良。腎、淋巴結、心內膜、心瓣膜及血管壁內均含有包涵體大細胞。腦組織神經元正常,無膠質變性僅有某些脂褐質顆粒,此與MPS Ⅰ型改變明顯不同。但腦灰質結締組織內有脂肪軟骨細胞營養不良浸潤。可在皮膚、黏膜和結膜內見到包涵體及膠原纖維異常。
臨床表現
臨床症狀與MPS I-H型相似,但較輕。最早出現和主要症狀是進行性角膜周邊部渾濁。可發生色素性視網膜炎。於6~8歲出現輕度關節強直,爪狀手,面、容輕度粗笨樣,口寬大多毛,無明顯侏儒。可有肝大,脾不大可有耳聾。智力正常,有的甚至聰明過人。也有的出現精神病症狀由於膠原組織壓迫神經,常出現腕管綜合徵(正中神經分布區感覺障礙,手指水腫,魚際萎縮),以後可出現主動脈瓣關閉不全或狹窄。
併發症:
本病可並發肝大,耳聾,精神病症狀,主動脈瓣關閉不全或狹窄,神經炎,角膜渾濁。
診斷
本型診斷與Ⅰ型相似,但無智力低下,發病年齡也較晚,並且有神經炎,角膜渾濁為周邊性。
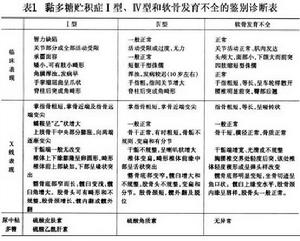
鑑別診斷:
關於本型的鑑別診斷,也與Ⅰ型相同。必須與其他幾型黏多糖貯積症仔細鑑別,尤其多糖貯積症Ⅳ型、軟骨發育不全仔細鑑別(表1)。
檢查
實驗室檢查:
尿中只出現硫酸皮膚素。血中白細胞及骨髓細胞內異染顆粒缺如或不清。
其它輔助檢查:
X線檢查。
X線表現與Ⅰ型相似,但輕得多,而且改變出現也較晚侏儒少見。
治療
目前無有效治療方法曾進行過各種試驗治療,但效果不肯定。糖皮質激素在鼠的實驗中有抑制黏多糖合成作用,但病人每天服用潑尼松2mg/kg並未出現臨床效果。大量維生素A加入MPSI-H和Ⅱ型病人培養的成纖維細胞可以使其異染性消失。給病人口服維生素A 1萬~10萬U,4次/d,經1個月~數月後可改善症狀,可使尿中黏多糖排出增多,但與體內沉積的黏多糖量相比則微不足道。維生素C的套用也未見臨床療效甲狀腺素或生長激素的套用也同樣無效。對於輸入新鮮血漿的治療方法意見尚有分歧,一部分作者認為可得到明顯的、暫時的臨床好轉,但其他作者套用卻未得到確切的療效。
預後預防
預後:
良好壽命似不受影響有活到60歲以上者。
預防:
糖原累積病是一組兒童遺傳性糖原代謝紊亂的疾病主要以機體組織糖原累積過多而分解困難為特點極少為糖原合成代謝障礙,致使機體組織糖原儲存少。糖原累積病並非為一種疾病,而是一組疾病。目前確定的已有12種。臨床均以低血糖為特徵,所涉及到的器官主要為肝、腎、骨骼肌。遺傳方式多為常染色體隱性遺傳,無性別差異,多在兒童期發病。部分病人至成人後,病情不再發展,可以維持一般健康水平。
患者主要是由於缺乏分解糖原的某些酶,如葡萄糖-6-磷酸酶、α-1,4葡萄糖甙酶、磷酸果糖激酶、肝磷酸化激酶等。
許多患者的父母為近親結婚,避免近親結婚是預防本病的重要環節。一旦發現糖原累積症,以防治低血糖為主,膳食少量多餐,限制脂肪和總熱量限制體力活動。血清乳酸高者,宜服碳酸氫鈉防治酸中毒。皮質激素、腎上腺素、胰高血糖素等可幫助控制低血糖。
