
病症簡介
骨髓增生異常綜合徵(myelodysplasticsyndrome,MDS)是一組起源於造血髓系定向幹細胞或多能幹細胞的異質性克隆性疾患,其基本病變是克隆性造血乾、祖細胞發育異常(dysplasia),導致無效造血以及惡性轉化危險性增高,主要特徵是無效造血和高危演變為急性髓系白血病,臨床表現為造血細胞在質和量上出現不同程度的異常變化,MDS發病率約10/10萬~12/10萬人口,多累及中老年人,50歲以上的病例占50%~70%,男女之比為2:1。MDS30%~60%轉化為白血病,其死亡原因除白血病之外,多數由於感染、出血,尤其是顱內出血。
疾病原因
發病原因
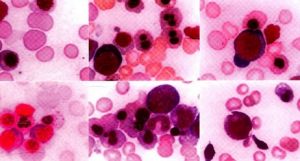 骨髓增生異常綜合徵
骨髓增生異常綜合徵繼發性MDS患者常有明顯發病誘因,苯類芳香烴化合物,化療藥物尤其是烷化劑,放射線均可誘導細胞基因突變而導致MDS或其他腫瘤發生,此外,MDS多發生於中老年,是否年齡可降低細胞內修復基因突變功能亦可能是致病因素之一。
發病機制
MDS患者在致病因素作用下,引起患者造血幹細胞損傷,用G6PD同工酶類型,X染色體伴限制性長度片段多態性甲基化,X染色體失活分析等方法已確定大部分MDS是病變發生在造血幹細胞水平的克羅恩病,因而不但髓系,紅系,巨核系細胞受累,淋巴細胞系亦受影響,導致T,B細胞數量和功能異常,臨床表現為免疫缺陷或自身免疫性疾病,但在部分患者中其發病可僅局限在粒,紅,巨核,巨噬祖細胞水平,僅有粒,紅,巨核,巨噬細胞等受累而無淋巴細胞受累。MDS發病具有階段特性,可能與不同原癌基因和抑癌基因的變化有關,原癌基因活化包括基因過量表達,擴張,重排,易位,點突變等,抑癌基因變化包括等位基因丟失,缺失,重排,突變,表達下降等,造血幹細胞在不同的增生分化階段受不同的原癌基因和抑癌基因調控,這種調控是通過其表達產物如生長因子,細胞表面受體,酪氨酸激酶類,ATP,胞質蘇氨酸/絲氨酸類,核蛋白類等完成,這些表達產物按嚴格的程式直接參與細胞增生分化的各個生理步驟,如某一生理環節由於原癌基因或抑癌基因調控失常,會引起細胞增生分化的紊亂,導致MDS或其他疾病。
在MDS發病初期某些有原癌基因或抑癌基因變化的造血幹細胞雖然伴有自身增生分化功能的某種異常,但仍可長期處於相對穩定階段,此時患者臨床病情穩定,僅有輕度貧血,白細胞,血小板減少,但當這一異常克隆進一步進展惡化時,此克隆衍生而來的另一種伴有染色體畸變的亞克隆幹細胞作為主要造血幹細胞來代替造血,染色體畸變使這一幹細胞有更明顯的增生分化異常,生成的各系不同階段血細胞常常不能分化成熟,中途凋亡比例增加,使外周血3系血細胞進一步減少,反饋刺激骨髓異常造血幹細胞加強增生,形成骨髓過度增生伴有病態造血表現,過度增生的異常克隆造血幹細胞常有兩種演變途徑:一為由於過度增生逐漸演變為造血能力衰亡,骨髓可轉為增生低下,臨床表現為造血功能衰竭,為半數以上MDS患者死亡原因,另一種演變為急性白血病,由MDS轉變為急性白血病大多為急性髓系細胞性白血病,僅極少數為急性淋巴細胞白血病,化療效果差,常不易緩解,即使緩解,緩解期也短。
症狀表現
多見貧血
骨髓增生異常綜合症的臨床表現無特殊性,此病患者通常起病緩慢,少數患者具有起病急劇的特點,一般從發病開始轉化為白血病,此病患者在一年之內約有50%以上會轉化為白血病,其中有貧血患者占90%,此類型患者就會有面色蒼白、乏力、活動後心悸、氣短等特點;當老年人出現貧血後會使原有的慢性心、肺疾病加重,此時出現發熱的患者會占50%,其中原因不明性發熱占10%~15%,此時患者會表現為反覆發生的感染及發熱,其中感染部位會以呼吸道、肛門周圍和泌尿系最多。肢體部位出血
人們能夠提前知曉骨髓增生異常綜合症病人的症狀表現,就會在自身有很多部位出血的情況及時來醫院檢查治療,目前此病導致人們出血的患者就占20%,其中常見的出血部位包括呼吸道及消化道,有些患者也會有顱內出血,早期的出血症狀較輕,大多是皮膚黏膜出血、牙齦出血或鼻衄;而女性患者就會有月經過多等表現,一旦病情到了晚期患者就會有出血趨勢加重,而腦出血成為患者死亡的主要原因之一。脾、肝腫大
提醒大家脾肝腫大幾乎是每位骨髓增生異常綜合症患者都會出現的症狀,病人會偶爾發現左上腹有一腫塊,有人認為脾大程度與病程有關,脾肋下每1CM代表一年病程;而且由於脾大,患者經常有腹部飽滿或沉重壓迫的感覺,對脾觸之堅實,一般無壓痛;但如脾增大太快,人們就會因脾局部梗死而發生局部疼痛,甚至可以聽到摩擦音。容易感染
人們得上骨髓增生異常綜合症之後會因粒細胞減少和功能異常導致感染髮生,病情初期比較穩定,患者多無嚴重的感染與發熱,後期就比較容易合併感染;而且由於免疫力低下就會容引起潛在性膿瘍以及化膿性關節炎、結核、綠膿桿菌性結膜炎、壞疽等不常見的感染;而且黴菌感染在後期較普遍,敗血症也會成為疾病終末期的合併症和主要的死亡原因。實驗室檢查
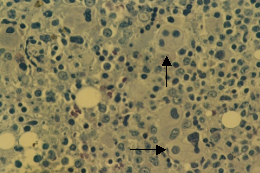 骨髓增生異常綜合徵
骨髓增生異常綜合徵外周血全血細胞減少,其程度依不同分型而異。如難治性貧血(RA)以貧血為主,難治性貧血伴有原始細胞增多(RAEB)或轉變中的RAEB(RAEB-T)則常有明顯的全血細胞減少。
二、骨髓象
大多數患者骨髓增生明顯或極度活躍,少數增生正常或減低。細胞形態異常反映了MDS的病態造血。紅系各階段幼稚細胞常伴類巨幼樣變,核漿成熟失衡,紅細胞體積大或呈卵園形,有嗜鹼點彩、核碎裂和Howell-Jolly小體。RA-S能檢出環形鐵粒幼細胞。粒系在RAEB和RAEB-T均可見原始細胞比例高於正常。粒細胞漿內顆粒粗大或減少,核分葉過多或過少,出現Pelger-Huët 畸形。部分胞漿內出現Auer小體。巨核細胞在數和質方面均可有異常,多數巨核細胞增多。檢出小巨核細胞是MDS的支持診斷指標之一。血小板體積大,顆粒少。
骨髓活檢在MDS已廣泛套用,不僅提供診斷依據,還有助於預測預後。骨髓病理切片中各系病態造血更加明顯,特別是粒系。若發現3~5個以上原粒與早粒聚集成簇,位於小梁間區或小梁旁區,即所謂“幼稚前體細胞異常定位”(abnormal localization of immature precursor,ALIP),是MDS骨髓組織的病理學特徵。凡ALIP陽性者,其向急性白血病轉化可能性大,早期死亡率高。反之,則預後較好。
三、細胞遺傳學研究
MDS是一種多能造血幹細胞水平上突變的獲得性克隆性疾病。過去,採用標準的染色體技術,31~49%原發性MDS患者中發現有某種染色體缺陷。近年,隨著染色體技術的改進,異常克隆的檢出率顯著提高。特異性染色體改變有-7/del7q,+8,-5/del5q,和累及第5、7和20號染色體的複合染色體異常。非特異性染色體改變,如環形染色體、雙著絲點染色體及染色體斷裂等。染色體的檢查對預測預後具有一定價值,骨髓中有細胞遺傳學異常克隆的患者,其轉化為急性白血病的可能性大得多,特別是-7/del7q和複合缺陷者,約72%轉化為急性白血病,中數生存期短,預後差。
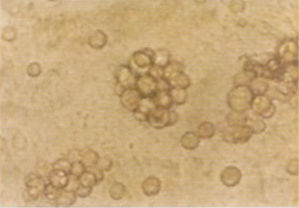 體外骨髓培養
體外骨髓培養MDS患者體外細胞培養中已發現的異常結果有:混合集落(CFU-GEMM)多不生長;原始細胞祖細胞(BCP)部分病例生長,部分不生長;粒細胞-單核細胞集落(CFU-GM)生成率減少;CFU-GM之叢落/集落比例增加;液體與軟瓊脂培養中成熟障礙;幼稚紅系祖細胞的爆式集落形成單位(BFU-E)和成熟紅系祖細胞的集落形成單位(CFU-E)生成率降低或不生長。上述變化隨著疾病進展,常可在體外骨髓培養中看到,如CFU-GM生成率進行性減少及叢落/集落比例逐漸增加的趨勢。體外培養的異常程度與向白血病轉化的可能性關係密切。
五、其他
約20%血清或尿溶菌酶升高。血清鐵蛋白有不同程度增加。約80%患者抗鹼血紅蛋白(HbF)輕、中度增加。獲得性HbH病可能是MDS的重要表現之一,HbH陽性者亦有較強的急性白血病轉化傾向。
診斷
1986年全國關於MDS的討論會提出下列診斷標準:骨髓至少兩系呈病態造血。外周血1系、2系或全血細胞減少,偶可白細胞增高,可見有核紅細胞或巨大紅細胞或其它病態造血現象。除外其他引起病態造血的疾病,如紅白血病、M2b型急性非淋巴細胞白血病、骨髓纖維化、慢性粒細胞白血病、原發性血小板減少性紫癜、巨幼細胞貧血等。
FAB小組將MDS分為五型,標準如下:
一、難治性貧血(RA)
血象:貧血,偶有的患者粒細胞減少、血小板減少而無貧血。網織紅細胞減少。紅細胞及粒細胞有病態造血現象。原始細胞無或<1%。骨髓:增生活躍或明顯活躍。紅系增生並有病態造血現象。很少見粒系及巨核系病態造血現象。原始細胞<5%。
二、環形鐵粒細胞性難治性貧血(RAS):
骨髓中環形鐵粒幼細胞數為骨髓所有有核細胞的15%以上,其他同RA。
三、難治性貧血伴有原始細胞增多(RAEB)
血象:2系或全血細胞減少,多見粒系病態造血現象,原始細胞<5%。
骨髓:增生明顯活躍、粒系及紅系都增生。3系都有病態造血現象,原始細胞Ⅰ型+Ⅱ型為5%~20%。
四、慢性粒,單核細胞白血病(CMML)
血象:單核細胞絕對值>1×109/L。粒細胞也增加並有顆粒減少或Pelger-Huet異常。原始細胞<5%。
骨髓:同RAEB,原始細胞5%~20%。
五、轉變中的RAEB(RAEB-T):血象及骨髓似RAEB,但具有下述三種情況的任一種:①血中原始細胞75%;②骨髓中原始細胞20~30%;③幼稚細胞有Auer小體。
六、了解臨床傷人病史,包括藥物最後和化學試劑的接觸史很重要,鑑別骨髓增生異常時尤其是原始細胞不高的病例,要考慮非克隆性疾病,若診斷困難可在幾個月後再行骨髓及細胞遺傳學檢查。
治療
支持治療
當患者有明顯貧血或伴心、肺疾患時,可輸紅細胞。RA和RA-S常因反覆輸血造成鐵負荷增加。在有出血和感染時,可輸入血小板和套用抗生素。預防性輸注粒細胞和血小板對MDS患者無明確療效。維生素治療
部分RA-S對維生素B6治療有效,200~500mg/日靜滴,可使網織紅細胞升高,輸血量減少。腎上腺皮質激素
約10~15%MDS患者,套用腎上腺皮質激素治療後,外周血細胞計數明顯上升,但皮質激素治療帶來的易感染,血糖升高等副作用不容忽視。分化誘導劑
MDS患者惡性克隆中的某些細胞仍保留分化潛能,一些藥物能誘導瘤細胞分化。目前常用的有1,25雙羥維生素D3,2μg/d口服,用藥至少12周。或用維生素D330~60萬單位肌注,每日一次,8~28周。在用藥中部分患者血象改善。該類藥物可引起威脅生命的嚴重高血鈣,故應嚴密監測血鈣變化。13-順式維甲酸在體外培養中有誘導分化作用,但臨床套用不理想,國內多採用全反式維甲酸20mg每日三次口服。小劑量阿糖胞苷對髓性白血病有分化誘導作用,目前已用於MDS,特別是RAEB和RAEB-T,緩解率約30%,10~20mg/m2/d皮下注射,7~21天。但小劑量阿糖胞苷對骨髓的抑制作用仍不能忽視,約15%患者死亡與藥物相關。雄激素
炔睪醇(danazol)是目前最常用的男性激素,600~800mgd,持續2~4月,但無確切療效。有報導認為男性激素有加速向急性白血病轉化的可能。聯合化療
就多數MDS而言,常規的抗白血病治療無益。MDS對化療耐受性低,治療療效差,即使獲得緩解,緩解期也短。若病人年齡小於50歲,處於RAEB-T臨床狀態好,可酌情用常規化療。骨髓移植
當年齡小於50歲,並處於RAEB或RAEB-T,有HLA同型供者,醫療條件允許,可考慮進行同種異體骨髓移植。預後
MDS是一種異質性疾病,各型間生存期差異較大。RA和RA-S患者生存期常>5年,CMML、RAEB和RAEB-T患者中數生存期常<1年。感染、出血及向AML轉化為主要死亡原因。
生活調理
 營養配餐
營養配餐2、忌口,雞屬陽,動風。MDS虛實夾雜,邪毒內壅助火動風之品宜忌。特別是陰虛火旺,出血,痰濕交阻者尤要注意。
3、冬蟲夏草燉鴨:冬蟲夏草5g,鴨75g,生薑3片,黃酒5g,水200ml,適加鹽油調味,文火燉兩小時,飲湯食肉。治療MDS氣陰不足,神疲乏力,舌淡紅,脈細者。
4、精神調理:肝氣鬱結與MDS的發病關係密切,有資料提出MDS發病前有長達半年以上的較嚴重的精神刺激,因此提倡虛懷若谷,胸襟開闊,提高修養,在疾病調製的過程中易非常關鍵。
