
反應過程
 鄰位效應
鄰位效應鄰位基團空間阻礙可以影響分子的對稱性,例如化合物a為手征性分子,而b卻為非手征性分子:
 圖1
圖1(圖1)
鄰位基團的空間阻礙可以把─NH2、─NO2等共軛基團排斥於苯環的共軛體系之外,從而使該化合物的鍵長、極性、酸鹼性等分子的靜態物理化學性能發生極大的變化。
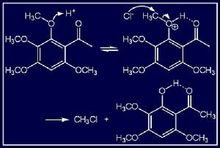
鄰位效應又如,由於氯原子的誘導效應,化合物e的酸性比化合物f、g為大。在化合物h、i中,由於化合物h是以分子間氫鍵為主,它的沸點可達295℃,而化合物i是以分子內氫鍵為主,它的沸點只有214℃。 鄰位效應對反應速率和反應機理也有影響。例如,
 圖3
圖3(圖3)化合物j能與碘甲烷反應生成四級銨鹽,但化合物k中由於鄰位上的兩個甲基的空間阻礙,─N(CH3)2基團平面偏離苯環平面,使化合物k極難形成四級銨鹽。 化合物 1在通常條件下的酯化速率要比化合物m、n慢得多。但在濃硫酸中,由於正碳離子的生成,進攻基團可以從與苯環垂直的方向進攻反應中心,所以化合物的酯化反應能順利地進行。這時的酯化機理已發生變化。
在某種反應過程里,相鄰基團部分地或完全和反應中心相鍵合,使的反應速率明顯增大,這種鄰位效應歸於鄰近基團的參與。因為鄰近基團參與,化合物p里的氯在沸水中易水解,可是化合物q在同樣條件下卻是穩定的。
因為鄰位基團的這種特殊的效應,鄰位基團對分子性能影響並不遵循哈米特方程。
取代反應
 鄰位效應
鄰位效應1、親核取代反應。簡稱SN。飽和碳上的親核取代反應很多。例如,鹵代烷能分別與氫氧化鈉、醇鈉或酚鈉、硫脲、硫醇鈉、羧酸鹽和氨或胺等發生親核取代反應,生成醇、醚、硫醇、硫醚、羧酸酯和胺等。醇可與氫鹵酸、鹵化磷或氯化亞碸作用,生成鹵代烴。鹵代烷被氫化鋁鋰還原為烷烴,也是負氫離子對反應物中鹵素的取代。當試劑的親核原子為碳時,取代結果形成碳-碳鍵,從而得到碳鏈增長產物,如鹵代烷與氰化鈉、炔化鈉或烯醇鹽的反應。由於反應物結構和反應條件的差異,SN有兩種機理,即單分子親核取代反應SN1和雙分子親核取代反應SN。
2、SN1的過程分為兩步:第一步,反應物發生鍵裂(電離),生成活性中間體正碳離子和離去基團;第二步,正碳離子迅速與試劑結合成為產物。總的反應速率只與反應物濃度成正比,而與試劑濃度無關。SN2為舊鍵斷裂和新鍵形成同時發生的協同過程。反應速率與反應物濃度和試劑濃度都成正比。能生成相對穩定的正碳離子和離去基團的反應物容易發生SN1,中心碳原子空間阻礙小的反應物容易發生SN2。如果親核試劑呈鹼性,則親核取代反應常伴有消除反應,兩者的比例取決於反應物結構、試劑性質和反應條件。低溫和鹼性弱對SN取代有利。
 鄰位效應
鄰位效應3、芳族取代反應。分芳族親電取代反應SEAr和芳族親核取代反應SNAr兩類,Ar表示芳基。芳烴通過硝化、鹵化、磺化和烷基化或醯基化反應,可分別在芳環上引進硝基、鹵原子、磺酸基和烷基或醯基,這些都屬SEAr。芳環上已有取代基的化合物,取代劑對試劑的進攻有定位作用。苯環上的取代基為給
電子基團和鹵原子時,親電試劑較多地進入其鄰位和對位;取代基為吸電子基團時,則以得到間位產物為主。此外,除發生這些正常反應外,有時試劑還可以進攻原有取代基的位置並取而代之,這種情況稱為原位取代。SNAr需要一定條件才能進行。如鹵代芳烴一般不易發生SNAr,但當鹵原子受到鄰或對位硝基的活化,則易被取代。鹵代芳烴在強鹼條件下也可發生取代。此外,芳香族重氮鹽由於離去基團斷裂成為穩定的分子氮,有利於生成苯基正離子,也能發生類似SNl的反應。
4、均裂取代反應。簡稱SH。為自由基對反應物分子中某原子的進攻,生成產物和一個新的自由基的反應。這種反應通常是自由基鏈式反應的鏈轉移步驟。一些有機物在空氣中會發生自動氧化,其過程也是均裂取代,如苯甲醛、異丙苯和四氫萘等與氧氣作用,可分別生成相應的有機過氧化物。
誘導效應
 鄰位效應
鄰位效應以取代苯苯環碳原子化學位移為依據,用曾經設定並初步證實的甲基誘導效應和共軛效應參數,討論了甲基取代基的誘導效應、共軛效應及定位效應。化學位移值表示的誘導效應參數表明甲基具有吸電子誘導效應,經校正後的化學位移值表示的共軛效應參數表明甲基具有微弱的給電子共軛效應,甚至其共軛效應有可能正處於給電子和吸電子的臨界狀態,總之相對於氫原子,甲基具有吸電子效應。甲基有較強的對位定位效應和較弱的鄰位定位效應,在適宜條件下間位也有可能發生取代反應。
場效應
 鄰位效應
鄰位效應直接通過空間和溶劑分子傳遞的電子效應。場效應是一種長距離的極性相互作用,是作用距離超過兩個C—C鍵長時的極性效應。場效應的作用方向與誘導效應作用方向往往相同,一般很難將這兩種效應區分開。R.戈爾登和L.M.斯托克測定了以下多環酸的電離常數[kg2]p:當X=H時,p為6.04;X=Cl時,p為6.25;X=COOCH時,p為6.20。在以上分子中,X基團與COOH基團之間相隔四個單鍵,X基團的誘導效應對p的變化影響很小。此外,分子內生成氫鍵的可能性也很小。因此,上述化合物的p的差別可用場效應來解釋。當X=Cl和COOCH時,其p大於X=H時的p,其原因是基團X吸收電子後形成偶極C—X的場效應,通過空間或溶劑分子直接影響了COOH的電離。場效應還與分子的幾何形狀有關。例如下列兩個化合物的p和酯化反應速率常數大小不一樣,它們的次序是a>b。在a、b兩個化合物中,氯原子對COOH的誘導效應是相同的,這兩種基團之間插入相同數目的C—C鍵。但它們的場效應則不相同,在化合物b中的氯原子通過空間與COOH的相互作用距離要比化合物a的作用距離近一些,因此a的酸度較大。這說明場效應確是存在的。
運用
苯甲酸運用:苯甲酸對蘑菇酪氨酸酶的單酚酶和二酚酶活力的影響以及抑制作用機理。研究結果表明,苯甲酸對蘑菇酪氨酸酶的單酚酶和二酚酶活性均有抑制作用,其效應為可逆抑制效應。測定導致單酚酶活力和二酚酶活力下降50%的抑制劑濃度(IC50)分別為1.20和1.00mmol/L。苯甲酸對蘑菇酪氨酸酶的單酚酶的遲滯時間有明顯的延長效應,4mmol/L苯甲酸使得單酚酶的遲滯時間從42s延長到200s。測定苯甲酸對二酚酶的抑制作用表現為非競爭性抑制類型,測定抑制常數為0.95mmol/L。苯甲酸與酶的結合導致天然酶的內源螢光的量子產率下降,但螢光發射峰沒有位移。作者提出抑制劑與酶分子的作用模型。
影響
 二氯苯
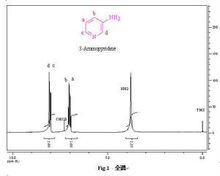
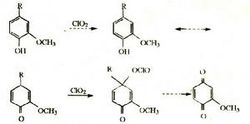
二氯苯質子影響:用鄰氯苯甲醛、對氯苯甲醛、鄰甲氧基苯甲醛、對甲氧基苯甲醛分別與環己酮縮二乙醇反應,合成了4種α,α'-雙亞苄基環烷酮類化合物。運用1H核磁共振手段,通過對雙鍵碳上H化學位移的分析,闡述了鄰位效應對質子化學位移的影響,認為是電子效應和空間效應等綜合作用的結果。鄰位基團的空間阻礙可以影響分子的對稱性,例如化合物a是手征性分子,而b卻是非手征性分子:
鄰位基團的空間阻礙可以把─NH、─NO等共軛基團排斥於苯環的共軛體系之外,從而使該化合物的鍵長極性、酸鹼性等分子的靜態物理化學性能發生極大的變化。例如化合物c中,由於硝基與對位氨基共軛,氨基上的孤電子對可轉移到硝基上去(c),它的偶極矩是6.18D。但化合物d中,由於鄰位甲基的空間阻礙,─NH和─NO基團平面偏離苯環平面,氨基上的電子對就不能轉移到硝基上去,它的偶極矩只有4.89D:
又如,由於氯原子的誘導效應,化合物e的酸性比化合物f、g為大。在化合物h、i中,由於化合物h是以分子間氫鍵為主,它的沸點可達295℃,而化合物i是以分子內氫鍵為主,它的沸點只有214℃。
鄰位效應對反應速率和反應機理也有影響例如,化合物j能與碘甲烷反應生成四級銨鹽,但化合物k中由於鄰位上的兩個甲基的空間阻礙,─N(CH)基團平面偏離苯環平面,使化合物k極難形成四級銨鹽。
化合物 1在通常條件下的酯化速率要比化合物m、n慢得多。但在濃硫酸中,由於正碳離子的生成,進攻基團可以從與苯環垂直的方向進攻反應中心,所以化合物的酯化反應能順利地進行。這時的酯化機理已發生變化。
在某種反應過程中,相鄰基團部分地或完全與反應中心相鍵合,使反應速率明顯增大,這種鄰位效應屬於鄰近基團的參與。例如,由於鄰近基團的參與,化合物p中的氯在沸水中易水解,但化合物q在同樣條件下卻是穩定的。
由於鄰位基團的這種特殊的效應,鄰位基團對分子性能的影響並不遵循哈米特方程。
