
研發歷程
2002年 阿昔替尼獲得特批進入實體腫瘤的臨床研究階段
2003年 阿昔替尼在歐美多國進入腎癌II期研究階段
2008年 輝瑞發起的多中心III期AXIS研究在歐美和亞洲開展
2012年 國際多中心AXIS研究顯示,阿昔替尼優於索拉非尼(NCT00678392)
2015年 中國臨床研究顯示,阿昔替尼優於索拉非尼
成份
本品主要成份為阿昔替尼。
化學名稱:N-甲基-2-[3-((E)-2-吡啶-2-基-乙烯基)-1H-吲哚-6-基磺醯]-苯甲醯胺
化學結構式:如右圖
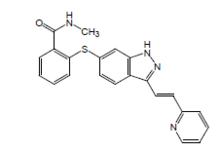 英立達
英立達分子式:CHNOS
分子量:386.47
性狀
1mg 片劑:紅色橢圓形薄膜衣片。
5mg 片劑:紅色三角形薄膜衣片。
適應症
阿昔替尼用於既往接受過一種酪氨酸激酶抑制劑或細胞因子治療失敗的進展期腎
細胞癌(RCC)的成人患者。
規格
(1)1mg;(2)5mg
用法用量
有腫瘤治療經驗的醫生才可使用阿昔替尼治療。
推薦劑量
阿昔替尼推薦的起始口服劑量為5mg(每日兩次)。阿昔替尼可與食物同服或在空腹條件下給藥,每日兩次給藥的時間間隔約為12 小時(見【藥代動力學】)。套用一杯水送服阿昔替尼。
只要觀察到了臨床獲益,就應繼續治療,或直至發生不能接受的毒性,該毒性不能通過合併用藥或劑量調整進行控制。
如果患者嘔吐或漏服一次劑量,不應另外服用一次劑量。應按常規服用下一次處方劑量。
劑量調整指南
建議根據患者安全性和耐受性的個體差異增加或降低劑量。
在治療過程中,滿足下述標準的患者可增加劑量:能耐受阿昔替尼至少兩周連續治療、未出現2 級以上不良反應(根據美國國立癌症研究所(NCI)不良事件常見術語標準[CTCAE])、血壓正常、未接受降壓藥物治療。當推薦從5mg BID 開始增加劑量時,可將阿昔替尼劑量增加至7mg BID,然後採用相同標準,進一步將劑量增加至10mg BID。
在治療過程中,一些藥物不良反應的治療可能需要暫停或永久中止阿昔替尼給藥,或降低阿昔替尼劑量(見【注意事項】)。如果需要從5mg BID 開始減量,則推薦劑量為3mg BID。如果需要再次減量,則推薦劑量為2mg BID。
合用CYP3A4/5 強效抑制劑:應避免合用CYP3A4/5 強效抑制劑(比如:酮康唑、伊曲康唑、克拉黴素、阿扎那韋、茚地那韋、奈法唑酮、奈非那韋、利托那韋、沙奎那韋、泰利黴素、伏立康唑)。建議選擇無CYP3A4/5 抑制潛能或有CYP3A4/5 微弱抑制潛能的藥物作為替代的合用藥物。儘管尚未在接受CYP3A4/5 強效抑制劑的患者中進行阿昔替尼劑量調整的研究,但如果必須與CYP3A4/5 強效抑制劑合用,建議將阿昔替尼的劑量減半,因為預計降低劑量後,阿昔替尼血漿濃度-時間曲線下面積(AUC)將調整至不與抑制劑合用的AUC 範圍內。可根據患者安全性和耐受性的個體差異增加或降低隨後劑量。如果停止與強效抑制劑合用,應將阿昔替尼劑量恢復至(當經過3-5 個抑制劑半衰期後)開始CYP3A4/5 強效抑制劑給藥前使用的劑量(見【藥物相互作用】和【藥代動力學】)。
合用強效CYP3A4/5 誘導劑阿昔替尼與強效CYP3A4/5 誘導劑合用可能降低阿昔替尼的血漿濃度(見【藥物相互作用】)。建議選擇無或僅有最低程度CYP3A4/5 誘導可能性的藥物作為替代的合用藥物。
儘管尚未在接受強效CYP3A4/5 誘導劑的患者中研究阿昔替尼劑量調整,但如果必須與強效CYP3A4/5 誘導劑合用,建議逐漸增加阿昔替尼的劑量。據報導,大劑量強效CYP3A4/5 誘導劑的最大誘導作用在合用該誘導劑治療一周內出現。如果阿昔替尼的劑量增加,應仔細監測患者的毒性。一些不良藥物反應的治療可能需要暫停或永久中止阿昔替尼治療,和/或降低阿昔替尼的劑量(見【注意事項】)。如果停止與強效誘導劑合用,應立即將阿昔替尼的劑量恢復至開始強效CYP3A4/5 誘導劑給藥前使用的劑量(見【藥物相互作用】)。
特殊人群
老人用藥:老年患者無需調整劑量(見【老年用藥】和【藥代動力學】)。
腎損害:目前尚未進行阿昔替尼在腎損害患者中的試驗。根據群體藥代動力學分析結果,觀察到阿昔替尼在輕度至重度腎損害患者中的清除率無顯著差異(15 mL/min≤肌酐清除率[CLcr] <89 mL/min)(見【藥代動力學】)。輕度至重度腎損害患者無需調整阿昔替尼起始劑量。終末期腎病患者(CLcr <15 mL/min)應慎用本品。
肝損害:當輕度肝損害患者服用阿昔替尼時,無需調整起始劑量(Child-Pugh 分級:A 級)。根據藥代動力學數據,當基線肝功能中度肝損害患者服用阿昔替尼時,起始劑量應減半(Child-Pugh 分級:B 級)。可根據患者安全性和耐受性的個體差異增加或降低隨後劑量。目前尚未在重度肝損害(Child-Pugh 分級:C 級)患者中進行過阿昔替尼研究,不應在該人群中使用阿昔替尼(見【注意事項】和【藥代動力學】)。
兒童人群
尚未在兒童患者中研究阿昔替尼的安全性和有效性。
不良反應
因為臨床試驗在各種不同條件下實施,因此一種藥物在臨床試驗中觀察到的不良反應發生率不能與另一種藥物在臨床試驗中觀察到的不良反應發生率進行直接比較,並且可能不能反映在臨床實踐中觀察到的不良反應發生率。
在單藥治療研究的715 例患者中評價了阿昔替尼的安全性,其中包括537 例晚期RCC 患者。所述數據反映了參加一項與索拉非尼相比的隨機臨床研究的359 例晚期RCC患者服用阿昔替尼的安全性(見【臨床試驗】)。
將在說明書的其他章節中更詳細地討論以下風險,包括應採取的相應措施(見【注意事項】):高血壓、動脈血栓栓塞事件、靜脈血栓栓塞事件、出血、心力衰竭、胃腸穿孔和瘺管形成、甲狀腺功能不全、傷口癒合併發症、RPLS、蛋白尿、肝酶升高、肝損害和胎兒發育。
禁忌
對阿昔替尼或任何輔料過敏。
注意事項
應在開始阿昔替尼治療之前和治療期間定期監測下列特定安全性事件。
高血壓和高血壓危象
在接受阿昔替尼治療的RCC 患者對照臨床研究中,接受阿昔替尼治療的145/359例患者(40%)和接受索拉非尼治療的103/355 例患者(29%)報告高血壓。在接受阿昔替尼治療的56/359 例患者(16%)和接受索拉非尼治療的39/355 例患者(11%)中觀察到3/4 級高血壓。接受阿昔替尼治療的2/359 例患者(<1%)報告高血壓危象,接受索拉非尼治療的患者未報告高血壓危象。高血壓(收縮壓>150mmHg,或舒張壓>100mmHg)的中位發生時間在開始阿昔替尼治療後1 個月內,並且在開始阿昔替尼治療後4 天就觀察到血壓升高。可採用標準抗高血壓藥物治療高血壓。接受阿昔替尼治療的1/359 例患者(<1%)因高血壓停止阿昔替尼治療,接受索拉非尼治療的患者無因高血壓停止治療(見【不良反應】)。
在開始阿昔替尼治療前,應控制好血壓。應監測患者高血壓出現情況,同時按需給予標準抗高血壓藥物治療。如給予抗高血壓藥物治療後仍存在持續性高血壓,應降低阿昔替尼劑量。如果同時給予抗高血壓藥物並降低阿昔替尼劑量仍出現嚴重且持續性高血壓,應停用阿昔替尼,一旦患者血壓正常即重新開始給予較低劑量的阿昔替尼。如果出現高血壓危象證據,應考慮停藥。如果中斷阿昔替尼給藥,應監測接受抗高血壓藥物治療的患者是否出現低血壓(見【用法用量】)。
如果懷疑存在可能與輕至重度高血壓有關的可逆性後部腦白質病綜合徵(RPLS)(見下),則應考慮進行診斷性腦部磁共振成像。
動脈血栓栓塞事件
在臨床試驗中,已報告包括死亡在內的動脈血栓栓塞事件。在接受阿昔替尼治療的RCC 患者對照臨床試驗中,接受阿昔替尼治療的4/359 例患者(1%)和接受索拉非尼治療的4/355 例患者(1%)報告3/4 級動脈血栓栓塞事件。在接受阿昔替尼治療的359例患者中,有1 例患者(<1%)報告致死性腦血管意外。在接受索拉非尼治療的患者中未報告致死性腦血管意外(見【不良反應】)。
在阿昔替尼臨床試驗中,有17/715 例患者(2%)報告動脈血栓栓塞事件(包括一過性腦缺血發作、腦血管意外、心肌梗死、視網膜動脈閉塞),有2 例死亡繼發於腦血管意外。
有發生此類事件風險或此類事件病史的患者應慎用阿昔替尼。目前尚未在之前12個月內發生動脈血栓栓塞事件的患者中研究阿昔替尼。
靜脈血栓栓塞事件
臨床試驗已報告包括死亡在內的靜脈血栓栓塞事件。在接受阿昔替尼治療RCC 的患者對照臨床試驗中,接受阿昔替尼治療的11/359 例患者(3%)和接受索拉非尼治療的2/355 例患者(1%)報告靜脈血栓栓塞事件。接受阿昔替尼治療的9/359 例患者(3%)(包括肺栓塞、深靜脈血栓、視網膜靜脈閉塞、視網膜靜脈血栓)和接受索拉非尼治療的2/355 例患者(1%)報告3/4 級靜脈血栓栓塞事件。致死性肺栓塞見於1/359 例(<1%)接受阿昔替尼治療的患者,接受索拉非尼治療的患者無報告。在阿昔替尼臨床試驗中,有22/715 例患者(3%)報告靜脈血栓栓塞事件,其中2 例死亡繼發於肺栓塞。
存在這些事件風險或曾有這些事件病史的患者應慎用阿昔替尼。目前尚未在之前6個月內發生靜脈血栓栓塞事件的患者中研究阿昔替尼。
血紅蛋白或血細胞比容升高
阿昔替尼治療過程中可能發生血紅蛋白或血細胞比容升高,反映紅細胞總量增加(見【不良反應】)。紅細胞總量增加可能增加血栓栓塞事件的風險。
應在開始阿昔替尼治療前並在治療過程中定期監測血紅蛋白或血細胞比容。如果血紅蛋白或血細胞比容升高至高於正常水平,應根據常規對患者進行治療,將血紅蛋白或血細胞比容降低至可接受的水平。
出血
在接受阿昔替尼治療的RCC 患者對照臨床研究中,接受阿昔替尼治療的58/359 例患者(16%)和接受索拉非尼治療的64/355 例患者(18%)報告出血事件。接受阿昔替尼治療的5/359(1%)例患者和接受索拉非尼治療的11/355(3%)例患者報告3/4 級出血事件(包括腦出血、咯血、血尿、下消化道出血和黑便)。接受阿昔替尼治療的1/359(<1%)例患者(胃出血)和接受索拉非尼治療的3/355(1%)例患者報告致死性出血。
目前尚未在未經治療的腦轉移患者或近期內出現活動性胃腸道出血患者中研究阿昔替尼,不應在這些患者中使用阿昔替尼。如果出血事件需要藥物干預,應暫停阿昔替尼給藥。
心力衰竭
在一項阿昔替尼治療RCC 患者的對照臨床研究中,6/359 例(2%)接受阿昔替尼治療的患者和3/355 例接受索拉非尼治療的患者(1%)報告心力衰竭。2/359 例(1%)接受阿昔替尼治療的患者和1/355(<1%)例接受索拉非尼治療的患者報告3/4 級心力衰竭。2/359 例(1%)接受阿昔替尼治療的患者和1/355 例(<1%)接受索拉非尼治療的患者報告致死性心力衰竭。
在整個阿昔替尼治療過程中需監測心力衰竭的體徵或症狀。可能需要通過永久停用阿昔替尼控制心力衰竭。
胃腸穿孔和瘺管形成
在接受阿昔替尼治療的RCC 患者對照臨床研究中,接受阿昔替尼治療的1/359 例患者(<1%)報告胃腸穿孔,接受索拉非尼治療的患者未報告胃腸穿孔。在阿昔替尼臨床試驗中,5/715 例患者(1%)報告胃腸穿孔,包括1 例患者死亡。除胃腸穿孔患者外,還有4/715 例患者(1%)報告形成瘺管。
在阿昔替尼治療期間,應定期監測胃腸穿孔或瘺管形成的症狀。
甲狀腺功能不全
在接受阿昔替尼治療的RCC 患者對照臨床研究中,接受阿昔替尼治療的69/359 例患者(19%)和接受索拉非尼治療的29/355 例患者(8%)報告甲狀腺功能減退。接受阿昔替尼治療的4/359 例患者(1%)和接受索拉非尼治療的4/355 例患者(1%)報告甲狀腺功能亢進。在治療前促甲狀腺激素(TSH)<5μU/mL 的患者中,接受阿昔替尼治療的79/245 例患者(32%)及接受索拉非尼治療的25/232 例患者(11%)的TSH 升高至10 μU/mL 或10 μU/mL 以上(見【不良反應】)。
在開始阿昔替尼治療前應監測甲狀腺功能,在阿昔替尼治療期間應定期監測甲狀腺功能。應根據常規對甲狀腺功能減退和甲狀腺功能亢進進行治療,以維持甲狀腺功能正常狀態。
傷口癒合併發症
尚未開展阿昔替尼對傷口癒合影響的正式研究。
應在預定手術前至少24 小時停止阿昔替尼治療。應根據傷口是否完全癒合的臨床判斷,決定術後重新開始阿昔替尼治療的時間。
可逆性後部腦白質病綜合徵
在接受阿昔替尼治療的RCC 患者對照臨床研究中,接受阿昔替尼治療的1/359 例患者(<1%)報告可逆性後部腦白質病綜合徵(RPLS),接受索拉非尼治療的患者未報告RPLS(見【不良反應】)。在阿昔替尼其他臨床試驗中報告了兩例RPLS。
RPLS 是一種神經系統疾病,可能表現為頭痛、癲癇發作、昏睡、意識模糊、失明、其他視覺和神經系統紊亂。還可能出現輕度至重度高血壓。核磁共振成像是確認RPLS診斷所必需的。出現RPLS 的患者應停用阿昔替尼。曾出現過RPLS 的患者再次給予阿昔替尼治療的安全性未知。
蛋白尿
在接受阿昔替尼治療的RCC 患者對照臨床研究中,接受阿昔替尼治療的39/359 例患者(11%)和接受索拉非尼治療的26/355 例患者(7%)報告蛋白尿。接受阿昔替尼治療的11/359 例患者(3%)和接受索拉非尼治療的6/355 例患者(2%)報告3 級蛋白尿(見【不良反應】)。
在開始阿昔替尼治療前應監測尿蛋白,在阿昔替尼治療期間應定期監測尿蛋白。出現中度至重度蛋白尿的患者應降低阿昔替尼劑量或暫停使用阿昔替尼。
肝酶升高
在接受阿昔替尼治療的RCC 患者對照臨床研究中,兩個治療組中分別有22%的患者發生過所有級別的ALT 升高。阿昔替尼治療組中<1%的患者和索拉非尼治療組中2%的患者發生3/4 級ALT 升高事件。最常見的肝臟相關不良反應包括ALT、AST 和血膽紅素升高(見【不良反應】)。未觀察到ALT(>3 倍正常上限[ULN])和膽紅素(>2 倍ULN)同時升高。
在一項臨床劑量探索研究中,ALT(12 倍ULN)和膽紅素(2.3 倍ULN)同時升高被認為是與藥物相關的肝毒性,在接受阿昔替尼起始劑量20 mg 每日兩次(推薦起始劑量的4 倍)的1 例患者中觀察到該毒性。
在開始阿昔替尼治療前應監測ALT、AST 及膽紅素,並在整個治療期間定期監測這些參數。
肝損害
在阿昔替尼臨床研究中,中度肝損害(Child-Pugh B 級)受試者中阿昔替尼的全身暴露量大約比正常肝功能受試者高2 倍。中度肝損害患者(Child-Pugh B 級)接受阿昔替尼治療時,建議降低劑量。尚未在重度肝損害(Child-Pugh C 級)的患者中研究(見【用法用量】和【藥代動力學】),不應在這類患者中使用阿昔替尼。
妊娠
根據阿昔替尼的作用機制,孕婦服用阿昔替尼可能對胎兒造成致命傷害。目前尚未在妊娠婦女中進行阿昔替尼的充分和對照研究。在小鼠發育毒性研究中,當母體暴露量低於臨床推薦劑量的人體暴露量時,觀察到阿昔替尼有致畸性、胚胎毒性和胎兒毒性。
當接受阿昔替尼治療時,應建議育齡婦女避孕。如果在妊娠期間使用本品,或者如果接受本品的患者懷孕,應告知患者藥物對胎兒的潛在危害(見【孕婦及哺乳期婦女用藥】)。
乳糖
本品含有乳糖。患有罕見遺傳疾病包括半乳糖不耐受、Lapp 乳糖酶缺乏或葡萄糖-半乳糖吸收不良的患者不應服用本品。
對駕駛和使用機器能力的影響
阿昔替尼對駕駛和使用機器能力的影響很小。應將阿昔替尼治療過程中患者可能發生的事件例如頭暈和/或疲勞告知患者。
【孕婦及哺乳期婦女用藥】
孕婦用藥
目前尚無在孕婦中服用阿昔替尼的充分和對照的研究。根據其作用機制,當孕婦服用阿昔替尼時,可能導致對胎兒的傷害。在小鼠發育毒性研究中,當母體暴露量低於臨床推薦劑量的人體暴露量時,觀察到阿昔替尼有致畸性、胚胎毒性和胎兒毒性。因此,當擬接受阿昔替尼治療時,應建議患者避孕。如果本品在妊娠期間使用,或當患者接受本品治療時懷孕,應告知患者藥物對胎兒的潛在危害。
在交配前及第一周妊娠期間,將阿昔替尼灌胃給予雌性小鼠,每日給藥兩次,在所有試驗劑量組(≥15 mg/kg/劑量,是患者以推薦起始劑量給藥所得全身暴露量[AUC]的約10 倍)中,觀察到著床後損失率升高。在胚胎-胎仔毒性研究中,妊娠小鼠在臟器形成期間經灌胃接受劑量為0.15、0.5、1.5mg/kg/劑量的阿昔替尼(每日給藥兩次)。在1.5mg/kg/劑量組中(該劑量是以推薦起始劑量給予患者所得AUC 的約0.5 倍),當無母體毒性時觀察到的胚胎-胎仔毒性包括畸形(齶裂),在≥0.5 mg/kg/劑量組中(以推薦起始劑量給予患者所得AUC 的約0.15 倍),觀察到的胚胎-胎仔毒性為骨骼鈣化異常。
哺乳婦女用藥
目前尚不明確阿昔替尼是否經人乳汁排泄。由於大多數藥物經乳汁排泄,且哺乳嬰兒存在發生阿昔替尼嚴重不良反應的可能,因此應考慮藥物對母體的重要性,決定是否終止哺乳或停用藥物。
【兒童用藥】
尚未在兒童患者中研究阿昔替尼的安全性和有效性。
在阿昔替尼BID 經口給藥1 個月或更長時間的未成熟小鼠及犬中觀察到骨和牙齒的毒性。在≥15 mg/kg/劑量(約為推薦起始劑量給予患者所得系統暴露量[AUC]的6 倍、15 倍),觀察到小鼠和犬的包括生長板增厚的骨骼影響。在以≥5 mg/kg/劑量(約為推薦起始劑量給予患者所得AUC 的1.5 倍)每日兩次給藥的小鼠中,觀察到生長中的門牙出現異常(包括齲齒、牙齒咬合不正、破損和/或缺失)。尚未在年幼動物中進行有關兒科患者的其他潛在毒性的評估。
【老年用藥】
在接受阿昔替尼治療的RCC 患者對照臨床研究中,接受阿昔替尼治療的123/359例患者(34%)≥65 歲。儘管不能排除某些年長患者的敏感性較高,但在≥65 歲與65 歲以下患者間,總體上未觀察到阿昔替尼的安全性和有效性存在差異。
老年患者無需調整劑量(見【用法用量】和【藥代動力學】)。
藥物相互作用
體外數據顯示阿昔替尼主要經CYP3A4/5 代謝,少量經CYP1A2、CYP2C19 和尿苷二磷酸-葡萄糖醛酸基轉移酶(UGT)1A1 代謝。
CYP3A4/5 抑制劑
酮康唑是CYP3A4/5 的強效抑制劑,在健康志願者中以400 mg 每日一次的劑量給藥7 天,可使單次口服5mg 阿昔替尼的平均曲線下面積(AUC)升高2 倍,使Cmax升高1.5 倍。阿昔替尼與強效CYP3A4/5 抑制劑(例如酮康唑、伊曲康唑、克拉黴素、紅黴素、阿扎那韋、茚地那韋、奈法唑酮、那非那韋、利托那韋、沙奎那韋及泰利黴素)合用可能升高阿昔替尼血漿濃度。葡萄柚也可能升高阿昔替尼血漿濃度。建議選擇無或有最低程度CYP3A4/5 抑制可能性的藥物合用。如果必須與強效CYP3A4/5 抑制劑合用,建議調整阿昔替尼的劑量(見【用法用量】)。
CYP3A4/5 誘導劑
利福平是CYP3A4/5 的強效誘導劑,在健康志願者中以600 mg 每日一次的劑量給藥9 天,使單劑量5mg 阿昔替尼的平均AUC 降低79%,使Cmax 降低71%。
阿昔替尼與強效CYP3A4/5 誘導劑(例如利福平、地塞米松、苯妥英、卡馬西平、利福布汀、利福噴汀、苯巴比妥及貫葉連翹[也稱作聖約翰草])合用可能降低阿昔替尼血漿濃度。建議選擇無或有最低程度CYP3A4/5 誘導可能性的藥物合用。如果必須與強效CYP3A4/5 誘導劑合用,建議調整阿昔替尼的劑量(見【用法用量】)。
CYP1A2 和CYP2C19 抑制劑
少量阿昔替尼(<10%)經CYP1A2 和CYP2C19 代謝。尚未研究這些同工酶的強效抑制劑對阿昔替尼藥代動力學的影響。由於這些同工酶的強效抑制劑可能會增加阿昔替尼血漿濃度,因此應慎用。
CYP 和UGT 抑制和誘導的體外研究
體外研究表明治療血漿濃度下,阿昔替尼不抑制CYP2A6、CYP2C9、CYP2C19、CYP2D6、CYP2E1、CYP3A4/5 或UGT1A1。
體外研究表明阿昔替尼可能抑制CYP1A2。因此,阿昔替尼與CYP1A2 底物合用可能導致CYP1A2 底物(例如茶鹼)血漿濃度升高。
體外研究還表明阿昔替尼可能抑制CYP2C8。然而,阿昔替尼與紫杉醇(一種已知的CYP2C8 底物)合用,沒有導致晚期癌症患者的紫杉醇血漿濃度升高,表明缺乏臨床CYP2C8 抑制。
人肝細胞的體外研究還表明阿昔替尼不誘導CYP1A1、CYP1A2 或CYP3A4/5。因此預期阿昔替尼聯合用藥不會降低合用的CYP1A1、CYP1A2 或CYP3A4/5 底物的體內血漿濃度。
P-糖蛋白的體外研究
體外研究表明阿昔替尼抑制P-糖蛋白。然而,預期在治療血漿濃度下阿昔替尼不會抑制P-糖蛋白。因此預期阿昔替尼聯合用藥不會增加體內地高辛或其它P-糖蛋白底物的血漿濃度。
【藥物過量】
尚無針對阿昔替尼藥物過量的治療。
在接受阿昔替尼治療的RCC 患者的一項對照臨床研究中,有1 例患者意外接受20mg 劑量,每日兩次持續治療4 天,出現頭暈(1 級)。
在一項阿昔替尼臨床劑量探索研究中,接受10mg BID 或20mg BID 起始劑量的受試者發生的不良反應包括高血壓、與高血壓相關的癲癇發作及致死性咯血。
在懷疑過量情況下,應停用阿昔替尼,同時給予支持性治療。
藥理毒理
藥理作用
阿昔替尼在治療劑量下可以抑制酪氨酸激酶受體,包括血管內皮生長因子受體(VEGFR-1、VEGFR-2 和VEGFR-3)。這些受體與病理性血管生成、腫瘤生長和癌症進展相關。體外試驗與小鼠體內模型試驗顯示阿昔替尼可抑制VEGF 介導的內皮細胞增殖與存活;在荷瘤小鼠模型中,阿昔替尼可抑制腫瘤生長及VEGFR-2 的磷酸化。
毒理研究
遺傳毒性:阿昔替尼Ames 試驗和人淋巴細胞染色體畸變試驗結果為陰性,小鼠骨髓微核試驗結果陽性。
生殖毒性:阿昔替尼可能損害人的生殖功能和生育力。在重複給藥毒性試驗中,小鼠經口給藥2 次/天,≥15 mg/kg/劑量(約為人推薦起始劑量時系統暴露(AUC)的7倍),或犬經口給藥2 次/天,≥1.5 mg/kg/劑量(約為人推薦起始劑量時系統暴露的0.1倍),可引起雄性動物睪丸/附睪重量降低、萎縮或退化、生髮細胞數量減少、精子減少或異常精子形成,精子密度或計數減少;在≥5 mg/kg/劑量(大鼠與犬分別是人推薦起始劑量時系統暴露的1.5 倍或0.3 倍)下可見雌性小鼠和雌性犬生殖道異常,包括性成熟延遲、黃體數減少或缺失、子宮重量降低和子宮萎縮。
小鼠生育力試驗中,雄性小鼠經口給藥2 次/天,至少70 天,阿昔替尼50mg/kg/劑量(約為人推薦起始劑量時系統暴露的57 倍)對雄性小鼠交配率與生育率未見影響。雌性小鼠經口給藥2 次/天,至少15 天,≥15mg/kg/劑量(約為人推薦起始劑量時系統暴露的10 倍)可引起雌性小鼠生育力及胚胎存活率降低。
在胚胎-胎仔毒性研究中,妊娠小鼠在臟器形成期間經口給予阿昔替尼,劑量為0.15、0.5、1.5mg/kg,2 次/天。1.5mg/kg 劑量(約為人推薦起始劑量時系統暴露量的0.5 倍)時可見胚胎-胎仔毒性,包括畸形(齶裂);在≥0.5mg/kg 劑量(約為人體推薦起始劑量的系統暴露量的0.15 倍)時可見骨骼鈣化異常。
藥代動力學
群體藥代動力學分析匯總了在健康受試者和癌症患者中完成的17 項試驗的數據。採用有一級吸收和滯後時間的雙室模型可充分描述阿昔替尼的濃度-時間曲線。
吸收和分布:以5mg 劑量單次口服給藥後,中位Tmax 範圍為2.5~4.1 小時。根據血漿半衰期,預計在給藥後2~3 天內達到穩態。與單次給藥相比,阿昔替尼以5mg 每日給藥兩次,導致藥物約1.4 倍蓄積。穩態時,阿昔替尼在1mg~20mg 劑量範圍內表現出線性藥代動力學。口服5mg 劑量後,阿昔替尼的平均絕對生物利用度為58%。
與空腹過夜服用相比,阿昔替尼與中等脂肪膳食同服,結果導致AUC 下降10%,與高脂肪、高熱量膳食一同給藥,結果導致AUC 升高19%。阿昔替尼可與食物同服或空腹給藥(見【用法用量】)。
阿昔替尼與人血漿蛋白高度結合(>99%),優先與白蛋白結合,與α1-酸性球蛋白的結合率適中。在晚期RCC 患者(n=20)中,在進食狀態下每日給予兩次5mg 劑量,Cmax 和AUC0-24 的幾何平均值(CV%)分別為27.8(79%)ng/mL、265(77%)ng.h/mL。清除率和表觀分布容積的幾何平均值(CV%)分別為38(80%)L/h、160(105%)L。
代謝和排泄:阿昔替尼的血漿半衰期範圍為2.5~6.1 小時。阿昔替尼主要經肝臟CYP3A4/5 代謝,少量經CYP1A2、CYP2C19、UGT1A1 代謝。當阿昔替尼以5mg 放射活性劑量口服給藥後,約41%放射活性從糞便中回收,有23%放射活性從尿液中回收。
阿昔替尼原形藥物(占給藥劑量的12%)是糞便中確認的主要成分;羧酸代謝產物和亞碸代謝產物占尿液中放射活性的絕大多數。在血漿中,N-葡萄糖醛酸代謝產物代表主要放射活性成分(占循環放射活性的50%),阿昔替尼原形藥物和亞碸代謝產物各占循環放射活性的20%。
與阿昔替尼相比,亞碸代謝產物和N-葡萄糖醛酸代謝產物作用於VEGFR-2 的體外效價降低400 倍或400 倍以上。
藥物-藥物相互作用
其他藥物對阿昔替尼的影響:阿昔替尼主要經肝臟CYP3A4/5 代謝。此外,阿昔替尼水溶性取決於pH 值,pH 值升高導致溶解度降低。CYP3A4/5 強效抑制劑、CYP3A4/5強效誘導劑和抗酸劑對阿昔替尼藥代動力學的影響見圖4(見【用法用量】和【藥物相互作用】)。
阿昔替尼對其他藥物的影響:體外研究顯示,阿昔替尼可能抑制CYP1A2 和CYP2C8。但是,阿昔替尼和紫杉醇(是一種CYP2C8 底物)合用並不升高患者的紫杉醇血漿濃度。
體外研究顯示,阿昔替尼在治療血漿濃度下並不抑制CYP2A6、CYP2C9、CYP2C19、CYP2D6、CYP2E1、CYP3A4/5 或UGT1A1。人肝細胞體外研究顯示,阿昔替尼也不誘導CYP1A1、CYP1A2 或CYP3A4/5。
在體外,阿昔替尼是外排轉運蛋白P-糖蛋白(P-gp)的一種抑制劑。但是,預計達到治療量血漿濃度的阿昔替尼並不抑制P-gp。
特殊人群中藥代動力學
兒童使用:尚未在18 歲以下患者中研究阿昔替尼。
肝損害:肝損害對阿昔替尼藥代動力學的影響見圖4(見【用法用量】和【注意事項】)。
腎損害:在590 例健康志願者和患者中完成了群體藥代動力學分析(根據事先存在的腎功能情況分析),其中包括5 例重度腎損害患者(15 mL/min ≤CLcr <29 mL/min)、64 例中度腎損害(30 mL/min ≤CLcr <59 mL/min)患者和139 例輕度腎損害(60 mL/min≤CLcr <89 mL/min)患者。輕度至重度腎損害對阿昔替尼藥代動力學無有意義的影響。僅獲得一例終末期腎病患者的數據(見【用法用量】)。
其他內在因素:人群藥代動力學分析結果顯示,患者年齡、種族、體重、體表面積、UGT1A1 基因型或CYP2C19 基因型對阿昔替尼的清除率無臨床相關性影響。
貯藏
保存於20°C~25°C。允許短期保存於15°C~30°C。
包裝
高密度聚乙烯瓶或鋁箔/鋁箔泡罩包裝。
包裝規格:
1mg 片劑:180 片/瓶;14 片/盒;28 片/盒;56 片/盒
5mg 片劑:60 片/瓶;14 片/盒;28 片/盒;56 片/盒
有效期
36 個月
執行標準
進口藥品註冊標準JX20130209
批准文號
進口藥品註冊證號:
(1)1mg 規格:H20150219,H20150220
(2)5mg 規格:H20150221,H20150222
