簡介
肌營養不良症(musculardystrophy,MD)是一組原發於肌肉的遺傳性疾病。
主要臨床特徵
為受累骨骼肌肉的無力和萎縮。
病因及危險因素
肌營養不良症的發病原因曾有血管源性、神經源性、肌纖維再生錯亂、肌細胞膜功能紊亂等學說。各種理論均有各自的支持點和不能解釋點。症狀表現
 肌營養不良症
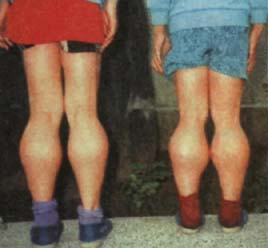
肌營養不良症(一)假肥大型呈性環連隱性遺傳,男性罹病,女性攜帶。通常在幼兒期起病。表現為能走路的年齡推遲,行走緩慢、易跌,跌倒後不易爬起。多數有小腿肌的肥大,病初肥大肌肌力相對稍強。臀中肌受累而致骨盆左右上下搖動;跟腱攣縮而足跟不能著地;腰大肌受累而腹部前凸,腦後仰。呈鴨型步態。從蹲位只有靠兩手撐著自己身體而逐步站直大腿,然後逐步挺起身子。繼骨盆帶肌肉受累之後,逐步出現肩胛帶 肌肉萎縮 、無力,兩臂舉高不能。菱形肌、前鋸肌、肩胛肌、崗上、崗下肌萎縮而使肩胛游離、肩胛骨呈翼狀聳起,稱翼狀肩。病程逐步發展,某些兒童可能由於本身生長發育的影響,出現病程的相對穩定或好轉。多數病孩在10歲時已喪失行走能力,依靠輪椅或坐臥不起,出現脊柱和肢體畸形。晚期,四肢攣縮,活動完全不能。常因伴發肺部感染、褥瘡等於20歲之前喪生。智商常有不同程度減退。半數以上可伴心臟損害,心電圖異常。早期呈現心肌肥大,除心悸外一般無症狀。
 肌營養不良症
肌營養不良症(二)面一肩一肱型呈常染色體顯性遺傳。男女均可罹病。病情嚴重程度不一,輕者可無任何主訴,在偶然機會或醫師進行家譜分析時發現。幼年或青春期隱匿起病,常在發病後數年才被引起注意。面肌受累較早,表現為 睡眠 時閉眼不緊、吹氣無力、苦笑臉容。逐步出現頸肌、肩胛帶肌、肱肌的萎縮、無力。肩胛帶和肱部肌肉萎縮,兩側肩峰隆突明顯。整個肩胛部酷似“衣架”。前臂肌肉正常。病程進展緩慢,軀幹和骨盆帶肌很晚累及。肢體遠端肌肉極少萎縮。偶伴腓腸肌肥大。多數病例不影響壽命。
(三)肢帶型較複雜,常為非單一疾病,有的呈常染色體隱性遺傳。 兩性 均可罹病。多數在青少年起病,個別更晚。以骨盆帶肌的無力、萎縮為首發症狀。進展緩慢,逐步累及肩胛帶而出現兩臂上舉困難、翼狀肩等典型症狀。晚期病者亦可出現肌肉攣縮、行動不能。無智慧型障礙。病情嚴重程度和進展速度差異很大,一般不影響壽命。
(四)眼肌型少見。部分病者是常染色體顯性遺傳。起病年齡不一。表現眼瞼下垂和進行性眼外肌麻痹。部分病例出現頭面部、咽喉部、頸部或(和)肢體肌肉無力和萎縮。少數病人可伴隨脊髓、小腦和視網膜受損,智慧型低下和腦脊液蛋白質異常增高。
(五)遠端型根據發病年齡自幼至中年後期不等亦可分為數種亞型,為常染色體顯性或隱性遺傳。表現為進行性遠端小肌肉萎縮。逐步向近端發展。進展極慢。不影響壽命。
診斷
根據起病隱匿、受累骨骼肌肉萎縮、無力的特殊分布和典型體徵,不難作出診斷。血清酶活性測定,肌電圖、肌肉活檢可為診斷和鑑別診斷提供佐證。某些類型的肌營養不良症仍需與多發性肌炎、運動神經元疾病或 重症肌無力 作鑑別。對DMD、BMD的確診有賴於在活檢肌肉發現dystrophin缺乏或異常,周圍血白細胞DNA分析發現dvstronhin基因變異。治療方法
至今尚無特效治療。潑尼松及硫唑嘌呤等免疫抑制劑效果不肯定。肌母細胞移植可緩解症狀,但因技術複雜而未能積極進行和推廣。
適當體育療法、按摩和跟腱延長等康復和 整形 手術仍是肌營養不良症病者最常用的治療方法。臥床不起者重視防上褥瘡和肺部感染。
中醫藥治療
中醫學認識:此病的主要原因為先天稟賦不足,或由於肝腎精虧,元氣不足,筋骨不充;或由於脾胃不健,肌肉失養而致。《諸病源侯論稱》:“脾與胃和,足陽明偉胃之經,胃為水谷之海也;脾主一身之肌肉,為胃消行水谷之氣,以養身體四肢。脾氣弱,即肌肉虛,受風邪所侵,故不能為胃通行水谷之氣,致四肢肌肉無所稟受。”本病病在肌肉,與肝腎脾及陽明經密切相關。病理性質為本虛標實,先天不足為本虛,痰濕壅滯為標實。
根據中醫理論,進行分型辨證治療。
(一) 脾腎兩虛(脾腎虛損)
主證:小腿或肩背肌肉假性肥大,四肢軟弱,不能抬舉,腰膝酸軟,步履乏力,手臂無力難以握持,肌肉萎縮,咀嚼無力,常有流涎,四肢不溫,大便溏薄,舌質淡胖,苔少,脈沉遲。
治法:補益脾腎,強肌健力。
代表方:右歸九合四君子湯加減。
(二)肝腎陰虛
主證:較晚學會走路,行走緩慢,鴨行步態,不能跑步絆跌,肌肉萎縮無力,頭暈耳鳴,腰膝酸軟,可有肌病面容,或小腿或肩背肌肉假性肥大,舌紅少苔,脈細數。
治法:滋補肝腎,強肌健力。
代表方:六味地黃湯加減。
(三)氣血虛弱
主證:肢體軟弱無力,手不能持重物,步履緩慢,起蹲困難,肌肉萎縮,最後可發生肢體攣縮,畸形。並有心悸氣短,面色蒼白無華,食少不化,舌體胖,舌質淡,苔薄白,脈沉細無力。
治法:益氣養血,強筋養肌。
代表方:滋血養筋湯加減。
