
簡介
分子幾何構型是分子結構和化學鍵理論的基本問題之一。了解分子幾何構型對於了解物質的性能與其內部結構的關係具有十分重要的意義。自1916年路易斯(Lewis)從電子觀點出發,首先提出由電子對引起化學鍵。這個電子理論廣泛地套用於闡明物質的分子結構。但是,這一理論根據經典靜電理論把電子看成是靜止不動的負電荷,必然會遇到許多不能解決的矛盾。1927年,海特勒(Heitter,W.)和倫敦(London,F)首先把量子力學理論套用到分子結構中。後來鮑林(Pauling,L.)等人發展了這一成果。建立了現代價鍵理論。1932年.密立根和洪特從不同於價鍵理論的另外角度提出了分子軌道理論。後來,為了更好地解釋分子的實際空間構型和穩定性,鮑林(Pauling,L.)在“電子配對”假設的基礎上又提出了“軌道雜化理論”。
幾何構型理論判斷
用理論來判定簡單多原子分子幾何構型的方法主要是在不同鍵長鍵角下計算分子的總能量,當總能量最低時所對應的鍵長鍵角即為分子的穩定幾何構型。
價鍵理論(VB理論)
該理論是用線性變分法處理H分子所得結果的推廣。當具有未成對電子數的原子接近時,未成對電子數以自旋反平行配對形成共價鍵,且共價鍵的形成將儘可能採取電子云最大重疊方向,即把分子中的電子先填人原子軌道,再考慮價電子在成鍵原子的原子軌道的交換來構成分子波函式,從而決定了分子的幾何構型。根據價鍵理論,為增加體系的穩定性,各原子價層軌道中未成對電子應儘可能相互配對,以形成最多數目的化學鍵。
例如:從分子N原子的基態電子組態為1S 2S 2P 2P 2P 有3個未成對電子,當2個N原子互相接近時,這些電子能兩兩配對形成共價三鍵,所以從分子具有三重鍵。
VB理論比較直觀,易於接受,對描述電子對鍵特別方便,但VB理論的計算部分較複雜,且在解釋單電子鍵,三電子鍵以及順磁性分子,共扼分子等結構時遇到很大困難。
分子軌道理論(MO理論)
MO理論是用線性變分法處理H所得結果的推廣。把分子軌道近似用適當的原子軌道的線性組合來表示,組合係數由變分法確定,組合得到的分子軌道數與參加組合的原子軌道數相同,分子中的電子排布遵循泡利原理,能量最低原理,洪特規則,參與組成分子軌道的原子軌道還必須滿足能量近似原理,軌道最大重疊原理和對稱匹配性條件,才能有效的組成分子軌道。
比較VB理論和MO理論,MO理論著重於分子的整體性,內層電子實際不參與成鍵,分子中各個分子軌道都具有一定的分布和能級,非常適合於描述分子的基態和激發態間的性質,了解各個狀態的波函式的分布和能級高低,闡明各種類型的分子光譜的性質,計算結果較VB理論精確,可見比VB理論的分析要深刻的多。
雜化軌道理論
該理論是VB理論的延伸。例如:實驗證明CH為正四面體分子構型,根據VB理論,C原子的電子組態為1S 2S 2P 2P 只有兩個未成對電子,理論上只能形成兩個C一H鍵,與實際不吻合。
 圖(1)常見雜化軌道
圖(1)常見雜化軌道若一個2S電子激發到2P軌道上,C(1S 2S 2P 2P )— C*(1S 2S 2P 2P 2P )就有四個未成對電子,可形成四個C一H鍵,並且消除了P軌道和S軌道之間的差別,使四個C一H鍵等同,就可以解釋C場的分子軌道。
雜化軌道理論說明滿足對稱性匹配和能量近似兩個條件的原子軌道就可以雜化,滿足正交歸一性的雜化軌道成鍵能力大於未雜化軌道的成鍵能力,體系更穩定。知道雜化軌道的成分即可計算出軌道之間的夾角和相對成鍵能力,求出雜化後的分子軌道表達式並解釋共價鍵的方向性和飽和性,從而大致判斷分子的幾何構型。
雜化軌道圖象簡單,能較好地解釋許多僅用“電子配對法”不能說明的分子空間結構和穩定性的事實,進一步發展了VB理論。雜化軌道理論成功地解釋了分子的空間構型,但一個分子究竟採取哪種雜化有時情況還是難以確定,而且雜化軌道理論不能確切地說明分子的電離,光譜性質等與單個電子行為有關的問題,還須依賴MO理論來解決。
價層電子對互斥理論(VSEPR理論)
該理論對解釋許多非共扼多原子分子的幾何構型很有幫助,而且非常簡單易行。在AX型分子或基團中,假如中心原子A的價電子層不含d電子或僅含球形對稱分布的d ,d 電子,則其幾何構型完全由中心原子的價電子數決定。當中心原子A的周圍有價電子對(包括成鍵電子對和孤對電子對)時,為使價電子對間斥力最小,可將成鍵電子對和孤對電子對看作等距離的排布在同一球面上,形成規則的多面體形式,價電子對中心之間的距離應保持最遠,使其總斥力最小,體系能量降到最低。因此從其價電子對數就可以定性推測分子的大致構型以及鍵長和鍵角的規律性。
不足之處:
(1)該理論通常忽略了孤對電子和成鍵電子以及單鍵和重鍵的區別,而在了解分子鍵角問題時,這些問題不能忽略。
(2)當中心原子的價電子層不能被視為球面時,例如:對不是d ,d 的過渡元素的原子作中心原子的化合物,一般不能用這個理論來判斷分子構型。
(3)當分子的幾何形狀主要不是由價電子對數決定或價電子總數為奇數時,此理論判據無效。
影響構型的因素
孤對效應
分子中由於成鍵電子對同時受中心原子和配位原子兩個原子核的吸引,所以電子云比較緊縮,而孤電子對只受一個原子核的吸引,而且所占軌道離核較近,在核附近占有較大的空間,電子云較“肥大”。這樣孤電子對對鄰近的軌道中的電子的排斥作用強於成鍵軌道中的成鍵電子對。因此孤電子對傾向於彼此儘量遠離,並且壓擠成鍵電子對。
孤電子對效應一方面能幫助我們選擇分子的穩定構型,另一方面還能用來說明分子空間構型畸變的原因。例如CH、NH、HO系列中,孤電子對分別為0、1、2 鍵角逐漸變小分別為109.5°、107.5°、104.5°。其原因是孤對電子對數目增多,擠壓成鍵電子對所致。
重鍵效應
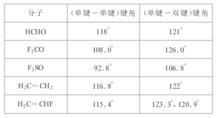 圖(2)含重鍵分子鍵角
圖(2)含重鍵分子鍵角重鍵軌道比單鍵軌道對其它軌道的排斥作用強。由於雙鍵中的四個電子或三鍵中的六個電子占據的空間大於單鍵中的兩個電子所占據的空間,對鄰近成鍵電子對的排斥力也大。因此含有重鍵的鍵角角度比較大,例如COCl分子中單鍵—雙鍵間鍵角為124.3°,而單鍵—單鍵鍵角為111.3°。
電負性效應
中心原子和配位原子間的電負性,也將影響分子的構型數據(P202- 205)。和中心原子鍵合的配位原子的電負性越大,吸引價電子的能力越強,價電子將向配位體方向移動,離中心原子較遠,減少了中心原子附近的電荷密度,從而使成鍵軌道與鄰近軌道間斥力減小。即價電子對間的排斥力隨配位原子的電負性增加而減少,生成的鍵角也較小。例如NF分子中,鍵角為102.1°;NH分子中,鍵角為107.5°。同理,配位原子相同,中心原子不同時,隨中心原子的電負性變小,則鍵角也減小。
但這一規則也有例外情況,如PF的鍵角(104°)> PX(X= Cl,Br,I)的鍵角。造成這種反常現象的原因,可能因為F是第二周期小原子,本身電荷密度已經很高,成鍵時P— F鍵的成鍵電子對又偏向於F原子,使F的電荷密度急劇增大。因此F原子可能將P或P電子反饋到P原子的d或d空軌道上,減少了F原子的電荷密度,增大了P原子的電荷密度,從而增大了P— F鍵間的斥力,使P— F鍵鍵角比較大。
相鄰電子對的數目
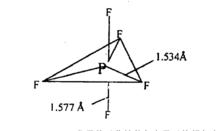 圖(3)
圖(3)價層電子對的相鄰電子對的數目對鍵長有影響。相鄰電子對數目多的價層電子對,受周圍其它電子對的排斥力大,離核較遠,即生成的鍵較長。例如AL分子每個豎立電子對都有3個成90°角的鄰對,而每個平狀電子對則有兩個成90°角和兩個成120°角的鄰對。因為成120°角的電子對間的斥力比成90°角的電子對間斥力小得多,所以豎立電子對所受總斥力大於平狀電子對,因而豎立鍵比平狀鍵長些,如圖(3)。
AL、AL、AL、AL分子的對稱性使各電子對的相鄰電子對的數目相同,因而這些類型分子的所有鍵長均相等。例如:COBClCClSF等分子就屬於這種類型分子。
