
基本概念
Southern印跡雜交是進行基因組DNA特定序列定位的通用方法。其基本原理是:具有一定同源性的兩條核酸單鏈在一定的條件下,可按鹼基互補的原則特異性地雜交形成雙鏈。一般利用瓊脂糖凝膠電泳分離經限制性內切酶消化的DNA片段,將膠上的DNA變性並在原位將單鏈DNA片段轉移至尼龍膜或其他固相支持物上,經乾烤或者紫外線照射固定,再與相對應結構的標記探針進行雜交,用放射自顯影或酶反應顯色,從而檢測特定DNA分子的含量 。
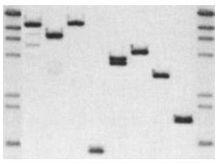 Southern印跡雜交顯色圖片
Southern印跡雜交顯色圖片由於核酸分子的高度特異性及檢測方法的靈敏性,綜合凝膠電泳和核酸內切限制酶分析的結果,便可繪製出DNA分子的限制圖譜。但為了進一步構建出DNA分子的遺傳圖,或進行目的基因序列的測定以滿足基因克隆的特殊要求,還必須掌握DNA分子中基因編碼區的大小和位置。有關這類數據資料可套用Southern印跡雜交技術獲得。
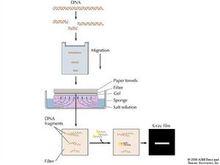 southern印跡雜交原理流程圖
southern印跡雜交原理流程圖Southern印跡雜交技術包括兩個主要過程:一是將待測定核酸分子通過一定的方法轉移並結合到一定的固相支持物(硝酸纖維素膜或尼龍膜)上,即印跡(blotting);二是固定於膜上的核酸與同位素標記的探針在一定的溫度和離子強度下退火,即分子雜交過程。該技術是1975年英國愛丁堡大學的E.M.Southern首創的,Southern印跡雜交故因此而得名。早期的Southern印跡是將凝膠中的DNA變性後,經毛細管的虹吸作用,轉移到硝酸纖維膜上。印跡方法如電轉法、真空轉移法;濾膜發展了尼龍膜、化學活化膜(如APT、ABM纖維素膜)等。利用Southern印跡法可進行克隆基因的酶切、圖譜分析、基因組中某一基因的定性及定量分析、基因突變分析及限制性片斷長度多態性分析(RFLP)等。
發明人
 埃德溫·邁勒·薩瑟恩
埃德溫·邁勒·薩瑟恩1975年,Southern印跡雜交(Southern blot)由英國人埃德溫·邁勒·薩瑟恩 (Edwin Mellor Southern)創建。
方法步驟
以哺乳動物基因組DNA為例,介紹Southern印跡雜交的基本步驟。
一、 待測核酸樣品的製備
(一)製備待測DNA
基因組DNA是從動物組織(或)細胞製備。1.採用適當的化學試劑裂解細胞,或者用組織勻漿器研磨破碎組織中的細胞;2.用蛋白酶和RNA酶消化大部分蛋白質和RNA;3.用有機試劑(酚/氯仿)抽提方法去除蛋白質。
(二) DNA限制酶消化
基因組DNA很長,需要將其切割成大小不同的片段之後才能用於雜交分析,通常用限制酶消化DNA。一般選擇一種限制酶來切割DNA分子,但有時為了某些特殊的目的,分別用不同的限制酶消化基因組DNA。切割DNA的條件可根據不同目的設定,有時可採用部分和充分消化相結合的方法獲得一些具有交叉順序的DNA片段。消化DNA後,加入EDTA,65℃加熱滅活限制酶,樣品即可直接進行電泳分離,必要時可進行乙醇沉澱,濃縮DNA樣品後再進行電泳分離。
二、 瓊脂糖凝膠電泳分離待測DNA樣品
(一)基本原理
Southern印跡雜交是先將DNA樣品(含不同大小的DNA片段)先按片段長短進行分離,然後進行雜交。這樣可確定雜交靶分子的大小。因此,製備DNA樣品後需要進行電泳分離。在恆定電壓下,將DNA樣品放在0.8~1.0%瓊脂糖凝膠中進行電泳,標準的瓊脂糖凝膠電泳可分辨70-80000bp的DNA片段,故可對DNA片段進行分離。但需要用不同的膠濃度來分辨這個範圍內的不同的DNA片段。原則是分辨大片段的DNA需要用濃度較低的膠,分辨小片段的DNA則需要濃度較高的膠。經過一段時間電泳後,DNA按分子量大小在凝膠中形成許多條帶,大小相同的分子處於同一條帶位置。另外為了便於測定待測DNA分子量的大小或是所處的分子大小範圍,往往同時在樣品鄰近的泳道中加入已知分子量的DNA樣品即標準分子量DNA (DNA marker)進行電泳。DNA marker可以用放射性核素進行末端標記,通過這種方式,雜交後的標準分子量DNA也能顯影出條帶。
(二) 基本步驟
1. 製備瓊脂糖凝膠,儘可能薄。DNA樣品與上樣緩衝液混勻,上樣。推薦使用的靶基因的上樣量見不同條件下上樣本的使用指針比較表。一般而言,對地高辛雜交系統,所需DNA樣品的濃度較低,每道加2.5-5μg人類基因組DNA;如果基因組比人類DNA更複雜(如植物DNA)則上樣量可達10μg;每道上質粒DNA<1ng。
2. 分子質量標誌物(DIG標記)上樣。
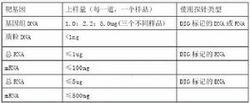 不同條件下上樣本的使用指針比較表
不同條件下上樣本的使用指針比較表3. 電泳,使DNA條帶很好的分離。4. 評價靶DNA的質量。在電泳結束後,0.25-0.50μg/mlEB染色15-30min,紫外燈下觀察凝膠。
三、電泳凝膠預處理
(一)原理
DNA樣品在製備和電泳過程中始終保持雙鏈結構。為了進行有效地實現Southern印跡轉移,對電泳凝膠做預處理十分必要。分子量超過10kb的較大的DNA片段與較短的小分子量DNA相比,需要更長的轉移時間。所以為了使DNA片段在合理的時間內從凝膠中移動出來,必須將最長的DNA片段控制在大約2kb以下。DNA的大片段必須被打成缺口以縮短其長度。因此,通常是將電泳凝膠浸泡在0.25mol/L 的HCl溶液短暫的脫嘌呤處理之後,移至於鹼性溶液中浸泡,使DNA變性並斷裂形成較短的單鏈DNA片段,再用中性pH的緩衝液中和凝膠中的緩衝液。這樣,DNA片段經過鹼變性作用,亦會使之保持單鏈狀態而易於同探針分子發生雜交作用。
(二)基本步驟
1. 如果靶序列>5kb,則需進行脫嘌呤處理。
(1) 把凝膠浸在0.25mol/L HCl中,室溫輕輕晃動,直到溴酚藍從藍變黃。
注意:處理人類基因組DNA≤10分鐘;處理植物基因組DNA≤20分鐘。
(2) 把凝膠浸在滅菌雙蒸水中
2. 如果靶序列<5kb,則直接進行下面的步驟
(1) 把凝膠浸在變性液(0.5mol/L NaOH,1.5mol/L NaCl)中,室溫2×15分鐘,輕輕晃動。
(2) 把凝膠浸在滅菌雙蒸水中
(3) 把凝膠浸在中和液中(0.5mol/L Tris-HCl,pH7.5,1.5mol/L NaCl),室溫2×15分鐘。
(4) 在20×SSC中平衡凝膠至少10分鐘。
四、轉膜
 各種尼龍膜性能及使用情況比較表
各種尼龍膜性能及使用情況比較表即將凝膠中的單鏈DNA片段轉移到固相支持物上。而此過程最重要的是保持各DNA片段的相對位置不變。DNA是沿與凝膠平面垂直的方向移出並轉移到膜上,因此,凝膠中的DNA片段雖然在鹼變性過程已經變性成單鏈並已斷裂,轉移後各個DNA片段在膜上的相對位置與在凝膠中的相對位置仍然一樣,故而稱為印跡(blotting)。用於轉膜的固相支持物有多種,包括硝酸纖維素膜(NC膜)、尼龍(Nylon)膜、化學活化膜和濾紙等,轉膜時可根據不同需要選擇不同的固相支持物用於雜交。其中常用的是NC膜和Nylon膜。各種膜的性能和使用情況比較見各種尼龍膜性能及使用情況比較表。
五、探針標記
用於Southern印跡雜交的探針可以是純化的DNA片段或寡核苷酸片段。探針可以用放射性物質標記或用地高辛標記,放射性標記靈敏度高,效果好;地高辛標記沒有半衰期,安全性好。人工合成的短寡核苷酸可以用T4多聚核苷酸激酶進行末端標記。探針標記的方法有隨機引物法、切口平移法和末端標記法。
六、預雜交(prehybridizafion)
 southern印跡雜交實驗儀器
southern印跡雜交實驗儀器將固定於膜上的DNA片段與探針進行雜交之前,必須先進行一個預雜交的過程。因為能結合DNA片段的膜同樣能夠結合探針DNA,在進行雜交前,必須將膜上所有能與DNA結合的位點全部封閉,這就是預雜交的目的。預雜交是將轉印後的濾膜置於一個浸泡在水浴搖床的封閉塑膠袋中進行,袋中裝有預雜交液,使預雜交液不斷在膜上流動。預雜交液實際上就是不含探針的雜交液,可以自制或從公司購買,不同的雜交液配方相差較大,雜交溫度也不同。但其中主要含有鮭魚精子DNA(該DNA與哺乳動物的同源性極低,不會與DNA探針DNA雜交)、牛血清等,這些大分子可以封閉膜上所有非特異性吸附位點。具體步驟如下:
(一) 配製預雜交液:6XSSC,5XDenhardt’s試劑,0.5%SDS,50%(v/v)甲醯胺,ddH2O,100ug/ml鮭魚精DNA變性後加入。註:1.每平方硝酸纖維素膜需預雜交液0.2ml。2.預雜交液製備時可用或不用poly(A)RNA。3.當使用32P標記的cDNA作探針時,可以在預雜交液或雜交液中加入poly(A)RNA以避免探針同真核生物DNA中普遍存在的富含胸腺嘧啶的序列結合。4.按照探針、靶基因和雜交液的特性確定合適的雜交溫度(Thyb)。(如果使用標準雜交液,靶序列DNA GC含量為40%,則Thyb為42℃。)
(二)把預雜交液放在滅菌的塑膠瓶中,在水浴中預熱至雜交溫度。
(三)將表面帶有目的DNA的硝酸纖維素濾膜放入一個稍寬於濾膜的塑膠袋,用5-10ml2×SSC浸濕濾膜。
(四)將鮭魚精DNA置沸水浴中10min,迅速置冰上冷卻1-2min,使DNA變性。
(五)從塑膠袋中除淨2×SSC,加入預雜交液,按每平方濾膜加0.2ml。
(六)加入變性的鮭魚精DNA置終濃度200μg/ml。
(七)儘可能除淨袋中的空氣,用熱封口器封住袋口,上下顛倒數次以使其混勻,置於42℃水浴中溫育4h.
七、Southern雜交
(一)原理
轉印後的濾膜在預雜交液中溫育4-6h,即可加入標記的探針DNA(探針DNA預先經加熱變性成為單鏈DNA分子),即可進行雜交反應。雜交是在相對高離子強度的緩衝鹽溶液中進行。雜交過夜,然後在較高溫度下用鹽溶液洗膜。離子強度越低,溫度越高,雜交的嚴格程度越高,也就是說,只有探針和待測順序之間有非常高的同源性時,才能在低鹽高溫的雜交條件下結合。
(二) 步驟
1.將標記的DNA探針置沸水浴10min,迅速置冰上冷卻1-2min,使DNA變性。
2.從水浴中取出含有濾膜和預雜交液的塑膠袋,剪開一角,將變性的DNA探針加到預雜交液中。
3.儘可能除取袋中的空氣,封住袋口,滯留在袋中的氣泡要儘可能地少,為避免同位素污染水浴,將封好的雜交袋再封入另一個未污染的塑膠袋內。
4.置42℃水浴溫育過夜(至少18h)。
八、洗膜
取出NC膜,在2XSSC溶液中漂洗5min,然後按照下列條件洗膜:2×SSC/0.1%SDS,42℃,10min,1S×SCC/0.1%SDS,42℃,10min,0.5S×SCC/0.1%SDS,42℃,10min,0.2×SSC/0.1%SDS,56℃,10min,0.1×SSC/0.1%SDS, 56℃,10min。採用核素標記的探針或發光劑標記的探針進行雜交還需注意的關鍵一步就是洗膜。在洗膜過程中,要不斷振盪,不斷用放射性檢測儀探測膜上的放射強度。當放射強度指示數值較環境背景高1-2倍時,即停止洗膜。洗完的膜浸入2×SSC中2min,取出膜,用濾紙吸乾膜表面的水分,並用保鮮膜包裹。注意保鮮膜與NC膜之間不能有氣泡。
九、放射性自顯影檢測
(一)將濾膜正面向上,放入暗盒中(加雙側增感屏)。
(二)在暗室內,將2張X光底片放入曝光暗盒,並用透明膠代固定,合上暗盒。
(三)將暗盒置-70℃低溫冰櫃中使濾膜對X光底片曝光(根據信號強弱決定曝光時間,一般在1-3天)。
(四)從冰櫃中取出暗盒,置室溫1-2h,使其溫度上升至室溫,然後沖洗X光底片(洗片時先洗一張,若感光偏弱,則在多加兩天曝光時間,再洗第二張片子)。(註:注意同位素的安全使用)
結果注意
在膜上陽性反應呈帶狀。實驗中應注意以下問題:轉膜必需充分,要保證DNA已轉到膜上。雜交條件及漂洗是保證陽性結果和背景反差對比好的關鍵。洗膜不充分會導致背景太深,洗膜過度又可能導致假陰性。若用到有毒物質,必需注意環保及安全。
主要套用
1.遺傳病診斷
2.DNA圖譜分析
3.檢測樣品中的DNA及其含量
4.PCR產物分析
