簡介
RT- PCR(Reverse Transcription-Polymerase Chain Reaction)即逆轉錄PCR,是將RNA的反轉錄(RT)和cDNA的聚合酶鏈式擴增(PCR)相結合的技術。首先經反轉錄酶的作用從RNA合成 cDNA,再以cDNA為模板,擴增合成目的片段。RT-PCR技術靈敏而且用途廣泛,可用於檢測細胞中基因表達水平,細胞中RNA病毒的含量和直接克隆 特定基因的cDNA序列。作為模板的RNA可以是總RNA、mRNA或體外轉錄的RNA產物。無論使用何種RNA,關鍵是確保RNA中無RNA酶和基因組 DNA的污染。
用於反轉錄的引物可視實驗的具體情況選擇隨機引物、Oligo dT 及基因特異性引物中的一種。對於短的不具有發卡結構的真核細胞mRNA,三種都可。
反轉錄酶的選擇
1. Money 鼠白血病病毒(MMLV)反轉錄酶:有強的聚合酶活性,RNA酶H活性相對較弱。最適作用溫度為37℃。
2. 禽成髓細胞瘤病毒(AMV)反轉錄酶:有強的聚合酶活性和RNA酶H活性。最適作用溫度為42℃。
3.Thermus thermophilus、Thermus flavus等嗜熱微生物的熱穩定性反轉錄酶:在Mn存在下,允許高溫反轉錄RNA,以消除RNA模板的二級結構。
4.MMLV反轉錄酶的RNase H突變體:商品名為SuperScript 和SuperScriptⅡ。此種酶較其它酶能多將更大部分的RNA轉換成cDNA,這一特性允許從含二級結構的、低溫反轉錄很困難的mRNA模板合成較長cDNA。
合成cDNA引物的選擇
1. 隨機六聚體引物:當特定mRNA由於含有使反轉錄酶終止的序列而難於拷貝其全長序列時,可採用隨機六聚體引物這一不特異的引物來拷貝全長mRNA。用此種方法時,體系中所有RNA分子全部充當了cDNA第一鏈模板,PCR引物在擴增過程中賦予所需要的特異性。通常用此引物合成的cDNA中96%來源於rRNA。
2. Oligo(dT):是一種對mRNA特異的方法。因絕大多數真核細胞mRNA具有3’端Poly(A)尾,此引物與其配對,僅mRNA可被轉錄。由於Poly(A)RNA僅占總RNA的1-4%,故此種引物合成的cDNA比隨機六聚體作為引物和得到的cDNA在數量和複雜性方面均要小。
3. 特異性引物:最特異的引發方法是用含目標RNA的互補序列的寡核苷酸作為引物,若PCR反套用二種特異性引物,第一條鏈的合成可由與mRNA 3’端最靠近的配對引物起始。用此類引物僅產生所需要的cDNA,導致更為特異的PCR擴增。
試劑準備
1.RNA提取試劑
2.第一鏈cDNA合成試劑盒
3.dNTPmix:含dATP、dCTP、dGTP、dTTP各2mM
4.Taq DNA聚合酶
操作步驟
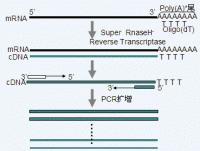 RT-PCR反應原理
RT-PCR反應原理1. 總RNA的提取:見相關內容。
2. cDNA第一鏈的合成:目前試劑公司有多種cDNA第一鏈試劑盒出售,其原理基本相同,但操作步驟不一。現以長沙贏潤生物技術有限公司提供的操作手冊為例:
(1)在0.5ml微量離心管中,加入總RNA 1-5μg,補充適量的DEPC H2O使總體積達11μl。在管中加10μM Oligo(dT)12-18 1μl,輕輕混勻、離心。
(2)70℃加熱10min,立即將微量離心管插入冰浴中至少1min。
然後加入下列試劑的混合物:
10×PCR buffer 2μl
25mM MgCl2 2μl
10mM dNTPmix 1μl
0.1M DTT 2μl
輕輕混勻,離心。42℃孵育2-5min。
(3)加入SuperscriptⅡ1μl ,在42℃水浴中孵育50min。
(4)於70℃加熱15min以終止反應。
(5)將管插入冰中,加入RNase H 1μl ,37℃孵育20min,降解殘留的RNA。-20℃保存備用。
3.PCR:
(1)取0.5ml PCR管,依次加入下列試劑:
第一鏈cDNA 2μl
上游引物(10pM) 2μl
下游引物(10pM) 2μl
dNTP(2mM) 4μl
10×PCR buffer 5μl
Taq 酶(2u/μl) 1μl
(2) 加入適量的ddH2O,使總體積達50μl。輕輕混勻,離心。
(3) 設定PCR程式。在適當的溫度參數下擴增28-32個循環。為了保證實驗結果的可靠與準確,可在PCR擴增目的基因時,加入一對內參(如G3PD)的特異性引物,同時擴增內參DNA,作為對照。
(4) 電泳鑑定:行瓊脂糖凝膠電泳,紫外燈下觀察結果。
(5) 密度掃描、結果分析:採用凝膠圖像分析系統,對電泳條帶進行密度掃描。
注意事項
1. 在實驗過程中要防止RNA的降解,保持RNA的完整性。在總RNA的提取過程中,注意避免mRNA的斷裂。
2. 為了防止非特異性擴增,必須設陰性對照。
3. 內參的設定:主要為了用於靶RNA的定量。常用的內參有G3PD(甘油醛-3-磷酸脫氫酶)、β-Actin(β-肌動蛋白)等。其目的在於避免RNA定量誤差、加樣誤差以及各PCR反應體系中擴增效率不均一各孔間的溫度差等所造成的誤差。
4. PCR不能進入平台期,出現平台效應與所擴增的目的基因的長度、序列、二級結構以及目標DNA起始的數量有關。故對於每一個目標序列出現平台效應的循環數,均應通過單獨實驗來確定。
5. 防止DNA的污染:
(1) 採用DNA酶處理RNA樣品。
(2) 在可能的情況下,將PCR引物置於基因的不同外顯子,以消除基因和mRNA的共線性。
