
範圍
GB 25572―2010 本標準適用於以石灰石或含石灰石的牡蠣殼等為原料經煅燒、消化而成的食品添加劑氫氧化鈣。
引用檔案
本標準中引用的檔案對於本標準的套用是必不可少的。凡是注日期的引用檔案,僅所注日期的版本適用於本標準。凡是不注日期的引用檔案,其最新版本(包括所有的修改單)適用於本標準。
分子式
Ca(OH)2
相對分子質量
74.09(按2007 年國際相對原子質量)
技術要求
感官要求:應符合表1 的規定。
表1 感官要求
| 項 目 | 要 求 | 檢驗方法 |
| 色澤 | 白色 | 取適量試樣置於50mL燒杯中,在自然光下觀察色澤和組織狀態。 |
| 組織狀態 | 粉末 |
理化指標:應符合表2 的規定。
表2 理化指標
| 項 目 | 指 標 | 檢驗方法 |
| 氫氧化鈣[Ca(OH)2],w/% | 95.0~100.5 | 附錄中氫氧化鈣的測定 |
| 碳酸鹽 | 通過試驗 | 附錄中碳酸鹽的測定 |
| 鎂及鹼金屬,w/% ≤ | 2.0 | 附錄中鎂及鹼金屬的測定 |
| 酸不溶物,w/ % ≤ | 0.1 | 附錄中酸不溶物的測定 |
| 砷(As)/(mg/kg)≤ | 2 | 附錄中砷的測定 |
| 氟化物(以F 計)/(mg/kg) ≤ | 50 | 附錄中氟化物的測定 |
| 鉛(Pb)/(mg/kg) ≤ | 2 | 附錄中鉛的測定 |
| 重金屬(以Pb 計)/(mg/kg)≤ | 10 | 附錄中重金屬(以Pb計)的測定 |
| 乾燥減量,w/% ≤ | 1.0 | 附錄中乾燥減量的測定 |
| 篩余物(0.045mm),w/% ≤ | 0.4 | 附錄中篩余物的測定 |
規範性附錄
檢驗方法
警示
本標準檢驗方法中使用的部分試劑具有腐蝕性,操作時須小心謹慎!如濺到皮膚上應立即用水沖洗,嚴重者應立即治療。使用劇毒品時,應嚴格按照有關規定管理;使用時應避免吸入或與皮膚接觸,必要時應在通風櫥中進行。暴露部位有傷口的人員不能接觸。
一般規定
本標準檢驗方法中所用試劑和水在沒有註明其他要求時,均指分析純試劑和GB/T 6682—2008 中規定的三級水。試驗中所用標準滴定溶液、雜質標準溶液、製劑及製品,在沒有註明其他要求時,均按HG/T3696.1、HG/T3696.2、HG/T3696.3 的規定製備。
鑑別試驗
試劑和材料
1 乙酸溶液:1+1。
2 草酸銨溶液:40g/L。
稱取4g 草酸銨(C2H8N2O4·H2O)溶於100mL 水中。
3 紅色石蕊試紙。
分析步驟
1 氫氧根離子的鑑別
稱取約5g 樣品,加入20mL 水混合,樣品形成稠糊,稠糊上層的澄清液使紅色石蕊試紙變藍。
2 鈣離子的鑑別
1g 樣品與20mL 水混合,加足量乙酸溶液使樣品溶解,加入草酸銨溶液,生成不溶的草酸鹽沉澱。此沉澱不溶於乙酸而溶於鹽酸。
氫氧化鈣的測定
方法提要
取適量試驗溶液,加入蔗糖掩蔽碳酸鹽的干擾,以酚酞為指示劑,用鹽酸標準滴定溶液滴定至無色。
試劑和材料
1 鹽酸標準滴定溶液:c(HCl)= 0.5mol/L。
2 蔗糖溶液:300g/L。
稱取300g 蔗糖,溶於1000mL 水中。加1 滴酚酞指示液, 使用前滴加氫氧化鈉溶液(4g/L)至溶液剛呈微粉色。
3 酚酞指示液:10g/L。
儀器和設備
電磁攪拌器。
分析步驟
稱取約0.5g 的試樣,精確至0.000 2g,置於250mL 錐形瓶中,加入50mL 水,振搖使之混勻。加入50mL 蔗糖溶液,用磁力攪拌器攪拌15min 後,加入2 滴~3 滴酚酞指示液,用鹽酸標準滴定溶液滴定至試液剛變為無色,並保持30s 不返色即為終點。同時做空白試驗,除不加試樣外,其他加入的試劑量與試驗溶液的完全相同(鹽酸標準滴定溶除外),並與試樣同時同樣處理。
結果計算
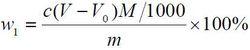 食品添加劑氫氧化鈣
食品添加劑氫氧化鈣氫氧化鈣含量以氫氧化鈣[Ca(OH)2]的質量分數w1 計,數值以%表示,按公式計算:
c——鹽酸標準滴定溶液濃度的準確數值,單位為摩爾每升(mol/L);
V——試驗溶液所消耗鹽酸標準滴定溶液體積的數值,單位為毫升(mL);
V0——空白試驗溶液消耗鹽酸標準滴定溶液體積的數值,單位為毫升(mL);
m——試樣的質量的數值,單位為克(g);
M——氫氧化鈣[1/2Ca(OH)2]摩爾質量的數值,單位為克每摩爾(g/mol)(M=37.05)。
取平行測定結果的算術平均值為測定結果,兩次平行測定結果的絕對差值不大於0.3%。
碳酸鹽的測定
試劑和材料
鹽酸溶液:1+3。
分析步驟
稱取約2g 試樣,精確至0.01g,加入50mL 水,混勻,加入40mL 鹽酸溶液,溶解過程應僅有細微的氣泡生成。
鎂及鹼金屬的測定
試劑和材料
1 硫酸。
2 鹽酸溶液:1+3。
3 氨水溶液:1+1。
4 草酸溶液:63g/L。
稱取6.3g 草酸(H2C2O4·2H2O)溶解在100mL 水中。
5 甲基紅指示液:1g/L。
儀器和設備
高溫爐:溫度可控制在800℃±25℃。
分析步驟
稱取約0.5g 樣品,精確至0.000 2g,加入10mL 水和6mL 鹽酸溶液使試樣溶解,煮沸1min。迅速加入40mL 草酸溶液,用力攪拌。加入2 滴甲基紅指示液,滴加氨水溶液,至溶液呈黃色,冷卻後將此混合液轉移到100mL 容量瓶中,用水稀釋至刻度,搖勻。靜置4h 或過夜。用中速濾紙乾過濾,棄去初始液10mL。用移液管移取50mL 濾液於已預先於800℃±25℃灼燒至質量恆定的瓷坩堝中,加入0.5mL 硫酸,水浴蒸發至近乾(或於電爐上低溫蒸發至近乾)。再在電爐上細心蒸發至乾。繼續加熱使銨鹽完全分解並揮發。置於高溫爐中,於800℃±25℃灼燒至質量恆定。
結果計算
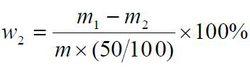 食品添加劑氫氧化鈣
食品添加劑氫氧化鈣鎂及鹼金屬含量以質量分數w2 計,數值以%表示,按公式計算:
m1——瓷坩堝和殘渣的質量的數值,單位為克(g);
m2——瓷坩堝的質量的數值,單位為克(g);
m—— 試料質量的數值,單位為克(g)。
取平行測定結果的算術平均值為測定結果,兩次平行測定結果的絕對差值不大於0.2%。
酸不溶物的測定
試劑和材料
1 鹽酸溶液:1+3。
2 硝酸銀溶液:17g/L。
儀器和設備
1 玻璃砂芯坩堝:孔徑5μm~15μm。
2 電熱恆溫乾燥箱:溫度可控制在105℃±2℃。
分析步驟
稱取約4g 試樣,精確至0.0002g,加少量水潤濕,加入60mL 鹽酸溶液使試樣溶解,加熱煮沸。用預先於105℃±2℃乾燥至質量恆定的玻璃砂芯坩堝趁熱過濾中,用熱水洗滌濾液至無氯離子(用硝酸銀溶液檢驗)。置於電熱恆溫乾燥箱中,於105℃±2℃乾燥至質量恆定。置於乾燥器中冷卻至室溫,稱量。
結果計算
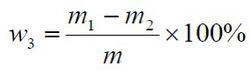 食品添加劑氫氧化鈣
食品添加劑氫氧化鈣酸不溶物含量以質量分數w3 計,數值以%表示,按公式計算:
m1——玻璃砂芯坩堝和殘渣的質量的數值,單位為克(g);
m2——玻璃砂芯坩堝的質量的數值,單位為克(g);
m—— 試料的質量的數值,單位為克(g)。
取平行測定結果的算術平均值為測定結果,兩次平行測定結果的絕對差值不大於0.03% 。
砷的測定
試劑和材料
1 鹽酸溶液:1+3。
2 其它試劑同GB/T 5009.76—2003 的第9 章。
儀器和設備
同GB/T 5009.76—2003 的第10 章。
分析步驟
稱取0.50g±0.01g 試樣,置於錐形瓶中。加入10mL 鹽酸溶液溶解試樣。加4mL 鹽酸,以下操作同GB/T 5009.76—2003 中第11 章“加水至30mL,再加5mL15%碘化鉀溶液……不得深於砷的限量
標準的砷斑。”
限量標準溶液的配製:移取1.00mL 砷標準溶液(1mL 溶液含砷1.00μg),以下操作同GB/T 5009.76—2003 中第11 章“加5mL 鹽酸……取出砷斑進行比較。”
氟化物的測定
試劑和材料
1 鹽酸溶液:1+11。
2 鹽酸溶液:1+3。
3 乙酸鈉溶液:3mol/L。
稱取204g 乙酸鈉(CH3COONa·3H2O),溶於300mL 水中,加乙酸溶液(1+16)調節pH 至7.0,加水稀釋至500mL。
4 檸檬酸鈉溶液:0.75mol/L。稱取110g 檸檬酸鈉(Na3C6H5O7·2H2O)溶於300mL 水中,加14mL 高氯酸,再加水稀釋至500mL。
5 總離子強度緩衝劑。乙酸鈉溶液與檸檬酸鈉溶液等量混合,使用前配製。
6 氟化物標準溶液:1mL 溶液含氟(F)0.010mg。移取1.00mL 按HG/T 3696.2 配製的氟化物標準溶液,置於100mL 容量瓶中,用水稀釋至刻度,搖勻。該溶液使用前配製。
儀器和設備
1 氟離子選擇電極。
2 飽和甘汞電極。
3 電磁攪拌器。
4 電位計:分度值為0.02。
分析步驟
1 儀器的準備
將氟離子選擇電極(按說明書活化)和飽和甘汞電極與測量儀器的負端與正端相連線,電極插入盛有水的50mL 塑膠杯中,在電磁攪拌中(使用聚乙烯轉子),讀取平衡電位值,更換2 次~3 次水後,待電位值平衡後,即可進行電位測定。
2 測定
稱取約1g 試樣,精確至0.01g。置於50mL 燒杯中,加水潤濕後,加入15mL 鹽酸溶液使樣品溶解。煮沸1min,冷卻後,轉移至50mL 容量瓶,加25mL 總離子強度緩衝劑,加水至刻度,搖勻。倒入50mL 塑膠燒杯中測定電極電位。
3 工作曲線的繪製
分別移取1.00mL,2.00mL,4.00mL,5.00mL,6.00mL 氟化物標準溶液(相當於含氟0.010mg,0.020mg,0.040mg,0.050mg,0.060mg)於5 只50mL 容量瓶中,於各容量瓶中分別加25mL 總離子強度緩衝劑,10mL鹽酸溶液,加水稀釋至刻度,搖勻。倒入50mL 塑膠燒杯中測定電極電位。以電極電位為縱坐標,氟的質量(mg)為橫坐標,在半對數坐標紙上繪製工作曲線,根據試樣的電位值在曲線上查得的氟的質量。
結果計算
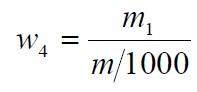 食品添加劑氫氧化鈣
食品添加劑氫氧化鈣氟化物含量以氟(F)的質量分數w4 計,數值以mg/kg 表示,按公式計算:
m1——根據測得的試驗溶液電位值從工作曲線上查得的氟的質量的數值,單位為毫克(mg);
m ——試料的質量的數值,單位為克(g)。
取平行測定結果的算術平均值為測定結果,兩次平行測定結果的絕對差值不大於5 mg/kg。
鉛的測定
石墨爐原子吸收分光光度法(仲裁法)
試劑和材料
1 硝酸。
2 硝酸溶液:0.5mol/L。將32mL 硝酸加入水中,稀釋至1000mL。
3 磷酸二氫銨溶液:20g/L。稱取2.0g 磷酸二氫銨,用水溶解,並稀釋至100mL。
4 鉛標準溶液:1mL 溶液含鉛(Pb)100ng。
移取1.00mL 按HG/T 3696.2 配製的鉛標準溶液置於100mL 容量瓶中,用硝酸溶液稀釋至刻度,搖勻。再用移液管移取5mL 稀釋過的溶液於500mL 容量瓶中,用硝酸溶液稀釋至刻度,搖勻。該溶液使用前配製。
5 二級水:符合GB/T 6682—2008 的規定。
儀器和設備
1 所用玻璃儀器:均以硝酸溶液(1+5)浸泡過夜,用水反覆沖洗,最後用去離子水沖洗乾淨。
2 原子吸收分光光度計(附石墨爐及鉛空心陰極燈)。
分析步驟
1 試驗溶液和空白試驗溶液的製備
稱取約0.5g 試樣,精確至0.01g,置於50mL 燒杯中,用水潤濕後,緩慢滴加約1.5mL 硝酸,低溫加熱,待樣品完全溶解後,升溫蒸至近乾,取下,冷卻至室溫,補加2 滴硝酸,轉移至25mL 容量瓶中,用水稀釋至刻度,搖勻。同時配製空白試驗溶液,此溶液除不加試樣外,其他加入試劑的種類和數量與試驗溶液相同,並同時操作。
2 工作曲線繪製
分別吸取鉛標準溶液0.00mL,5.00mL,10.00mL,20.00mL,30.00mL,40.00mL 於6 個50mL 的容量瓶中,用硝酸溶液稀釋至刻度,搖勻,此系列溶液濃度分別為0 ng/mL,10ng/mL,20ng/mL,40ng/mL,60ng/mL,80ng/mL。各吸取10μL,注入石墨爐,測得其吸光度,以鉛的濃度為橫坐標,對應的吸光度為縱坐標繪製工作曲線。
3 測定
在波長283.3nm 處將儀器調至最佳工作狀態,分別吸取試驗溶液和空白液10μL,注入石墨爐,測得其吸光度,從工作曲線上查得相應鉛的濃度。
4 基體改進劑的使用
若有干擾,則注入適量的基體改進劑磷酸二氫銨溶液,一般為5μL 或與試樣同量消除干擾。繪製鉛標準曲線時也要加入與試樣測定時等量的基體改進劑磷酸二氫銨溶液。
5 結果計算
鉛含量以鉛(Pb)的質量分數w5 計,數值以mg/kg 表示,按公式計算:
 食品添加劑氫氧化鈣
食品添加劑氫氧化鈣c1——從工作曲線上查出的試驗溶液中鉛的含量的數值,單位為納克每毫升(ng/mL);
c0 ——從工作曲線上查出的空白試驗溶液中鉛的含量的數值,單位為納克每毫升(ng/mL);
m ——試料的質量的數值,單位為克(g);
25——試料定容體積,單位為毫升(mL)。
取平行測定結果的算術平均值為測定結果,兩次平行測定結果的絕對差值不大於1 mg/kg。
雙硫腙分光光度法
試劑和材料
1 鹽酸溶液:1+3。
2 其它試劑同GB/T 5009.75—2003 中第3 章。
儀器和設備
同GB/T 5009.75—2003 中第4 章。
分析步驟
稱取約1g 試樣,精確至 0.01g,置於50mL 燒杯中。加水潤濕後,加入15mL 鹽酸溶液使試樣溶解,轉移至125mL 分液漏斗中,加1%硝酸溶液至20mL。同時作空白試驗,除不加試樣外,其他加入的試劑量與試驗溶液的完全相同,並與試樣同時同樣處理。以下同GB/T 5009.75—2003 中6.2“吸取鉛標準溶液……繪製工作曲線”。
結果計算
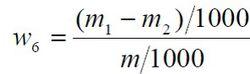 食品添加劑氫氧化鈣
食品添加劑氫氧化鈣鉛含量以鉛(Pb)的質量分數w6 計,數值以mg/kg 表示,按公式計算:
m1 ——從工作曲線上查出試驗溶液中鉛的質量的數值,單位為微克(μg);
m2 ——從工作曲線上查出空白試驗溶液中鉛的質量的數值,單位為微克(μg);
m ——試料的質量的數值,單位為克(g)。
取平行測定結果的算術平均值為測定結果,兩次平行測定結果的絕對值不大於1 mg/kg。
重金屬(以Pb計)的測定
試劑和材料
1 鹽酸溶液:1+1。
2 鹽酸溶液:1+3。
3 氨水溶液:2+3。
4 乙酸鹽緩衝溶液:pH﹦3.5。
稱取25.0 g 乙酸銨,加25 mL 水溶解,加45 mL 鹽酸溶液(A.11.1.1),再用鹽酸溶液或氨水溶液調節pH 值至3.5,用水稀釋至100 mL。
5 硫化鈉溶液;
稱取5 克硫化鈉,加10mL 水溶解,加入30mL 丙三醇,混勻,加蓋密封避光保存。配製後三個月內有效。
6 鉛標準溶液:1mL 溶液含鉛(Pb)0.01mg。
移取1.00mL 按HG/T 3696.2 配製的鉛標準溶液,置於100mL 容量瓶中,用水稀釋至刻度,搖勻。該溶液使用前配製。
7 酚酞指示液:10g/L。
儀器和設備
比色管:50mL。
分析步驟
稱取2.00g±0.01g 試樣,置於蒸發皿中,加水潤濕後,加入 30mL 鹽酸溶液使試樣溶解,水浴蒸至乾,加20mL 水使溶解,過濾於比色管中。加1 滴酚酞指示液,用氨水溶液調節至剛呈微粉色,加5 mL 乙酸鹽緩衝溶液,用水稀釋至刻度,加1 滴硫化鈉溶液,搖勻,於暗處放置5 min。在白色背景下觀察,所呈顏色不得深於標準比色溶液。
標準比色溶液的製備:移取2.00mL 鉛標準溶液於比色管中,加水至20mL,加5 mL 乙酸鹽緩衝溶液,用水稀釋至刻度,加1 滴硫化鈉溶液,搖勻,於暗處放置5 min。與試料同時處理。
乾燥減量的測定
儀器和設備
1 稱量瓶:Φ40×25mm。
2 電熱恆溫乾燥箱:溫度可控制在105℃±2℃。
分析步驟
稱取約2g 試樣,精確至0.0002g,置於預先於105℃±2℃下乾燥至質量恆定的稱量瓶中,置於電熱恆溫乾燥箱,在105℃±2℃下乾燥1h。取出,於乾燥器中冷卻至室溫,稱量。
結果計算
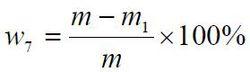 食品添加劑氫氧化鈣
食品添加劑氫氧化鈣乾燥減量以質量分數w7 計,數值以%表示,按公式計算:
式中:
m——乾燥前試料的質量的數值,單位為克(g);
m1 ——乾燥後試料的質量的數值,單位為克(g);
取平行測定結果的算術平均值為測定結果,兩次平行測定結果的絕對值不大於0.03%。
篩余物的測定
儀器和設備
1 試驗篩:R20/3 系列,Ф200×50—0.045/0.032 GB/T 6003.1 —1997。
2 軟毛刷。
分析步驟
稱取約10g 試樣,精確至0.01g。移入試驗篩內,用軟毛刷輕刷試樣,使粉末通過,最後,在篩子下墊一張黑紙,輕刷篩子直至所墊黑紙上沒有試樣痕跡。將篩余物轉移到已知質量的表面皿中稱量,精確至0.0002g。
結果計算
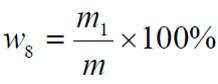 食品添加劑氫氧化鈣
食品添加劑氫氧化鈣篩余物含量以質量分數w8 計,數值以%表示,按公式計算:
m1——篩余物的質量的數值,單位為克(g);
m ——試料的質量的數值,單位為克(g)。
取平行測定結果的算術平均值為測定結果,兩次平行測定結果的絕對差值不大於0.04%。
