
簡介
化合物的結構、反應機制和合成三者的關係極其密切,相互依存。D.H.R.Barton在研究甾族化合物的反應時,在Hassel構象概念基礎上提出了構象分析的基本原理,指出了反應性與構象之間的聯繫。這是有機結構理論的一個突破性進展,也是動態立體化學的開始。反應機制與構象分析相結合,使許多反應的立體化學,即反應方向如區域選擇性和立體選擇性等可以預測,進而大大提高了有機合成設計的正確率。
環己烷衍生物分子的椅式構象中,取代基往往儘可能多地處於平伏鍵,以避免位於直鍵位所帶來的1,3-二直鍵相互作用。而E.J.Corey發現,構象剛性的環己烷類化合物分子,其環上碳原子受進攻時,成鍵位於直鍵位的產物反而占優勢。
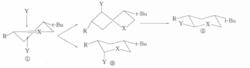 椅式構象
椅式構象例如,化合物①構象剛性,進攻試劑Y可從雙鍵所在平面的上方或下方垂直接近底物,從平面上方進攻需經歷扭船式中間體,而從平面下方無需經歷這樣的中間體,所以反應歷程中的過渡態或中間體的構象的能量差別是導致直鍵產物占優勢的直接原因。這種效應在有機化學文獻中名稱各異,有的稱為構象效應,有的稱為幾何制約,有的稱為構象最小改變原理。
舉例分析
分子由於構象不同而對分子的化學反應性質所產生的不同影響可統稱為構象效應。下面舉例加以說明。
例一
2,3一二溴丁烷非對映體,在反應上表現出不同的反應速度,如在碘化鉀一丙酮中的脫溴反應,內消旋體的反應速度比旋光異構體快1.8倍。該反應按E歷程進行,E反應對分子中被消除的兩個基團(此處為Br和Br)的立體化學要求是反式共平面。2,3-二溴丁烷內消旋體的優勢構象為對位交叉式,恰好符合E反應的立體化學要求,而且兩個較大的基團(一CH)距離遠,過渡態穩定。
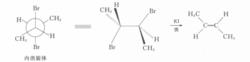 反應方程式
反應方程式在2,3-二溴丁烷旋光異構體的構象中,如能滿足兩個離去基團處於反式共平面的位置,則兩個較大基團(一CH)為鄰位交叉式,距離近,過渡態不穩定,故旋光異構體脫溴反應速度較慢。
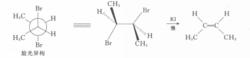 反應方程式
反應方程式而甲基被苯基替代後的脫溴反應速度,內消旋體比旋光異構體快100倍。這是因為苯基的體積較大,兩個大體積的苯基處於同側時過渡態更不穩定,需要更大的活化能。
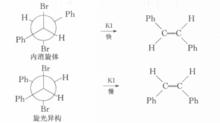 內消旋體/旋光異構體
內消旋體/旋光異構體例二
1,4-叔丁基環己醇,其反式乙醯化速度比順式醇的大3.7倍。
 1,4-叔丁基環己醇
1,4-叔丁基環己醇這是因為乙醯化反應速度取決於活化能的大小。活化能小的反應速度快,反式的活化能低,只有0.17kJ·mol 。同時,由於反式醇處於平伏鍵上,它比順式醇穩定,它的乙醯化中間體過渡態的張力要比直立鍵的同樣中間過渡態小。
構象效應與化學活性
最早指出構象與化學活性之間關係的是巴頓(Barton)。研究分子的構象不僅要了解分子的形狀及穩定構象,還要研究構象對反應活性的影響,尤其後者的研究是相當廣泛的。其中在環己烷體系中反應活性的構象效應曾被深入地研究過,對於發生反應的官能團,處於直立鍵上或是平伏鍵上其反應速度有明顯的差別。
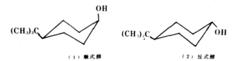 順式醇/反式醇
順式醇/反式醇順式醇羥基在直立鍵上,反式醇羥基在平伏鍵上,反式醇顯然比順式醇穩定。醯化反應的速度取決於活化自由能的大小,當乙醯基對羥基親電進攻時,由於反式醇的羥基處於平伏鍵上,它比順式醇穩定.因而反式醇乙醯化中間過渡態的張力要比順式醇的中間過渡態小些,反式的過渡態能量比順式的過渡態能量不止低2.90kJ/mol,活化能較小,由此可知反式醇乙醯化反應速度比順式醇快。如下圖所示:
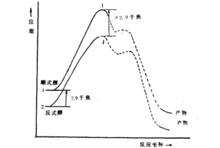 活化能
活化能順式醇之所以比反式醇氧化的速度快,也是由於反應的活化自由能不同的緣故。從中間體的結構來看,先是鉻酸與醇形成酯在C-H斷裂的同時消去一分子四價鉻化合物,由於直立式的相互排斥,所以順式酯的分解速度較反式酯的分解要快。它們在反應過程中的過渡態能量相差不到2.90k J/mol,速度相差約3倍。
