
發病機制
Ph染色體是慢性髓系白血病特徵性改變,由22號染色體的長臂與9號染色體的短臂相互易位的結果,即t(9;22)(q34;q11)。97.5%的Ph(+)CML具有典型的t(9;22)易位,其餘以變異Ph易位形式出現。易位的結果是形成BCR-ABL融合基因,產生BCR-ABL蛋白,有證據提示BCR-ABL蛋白可能直接產於白血病的形成。臨床表現
 慢性髓系白血病
慢性髓系白血病1.脾大
腫大程度不一,與外周血白細胞升高的水平有關;40%~70%的患者初診時脾臟在肋下10cm左右,通常無觸痛。加速期有與白細胞不成比例的脾臟迅速腫大伴壓痛。
2.發熱、貧血和出血
由於腫瘤負荷增加,可出現畏熱、消瘦和盜汗等高代謝綜合徵。早期因白細胞計數高而少有感染,明顯的貧血及出血多在急變期出現。
3.白細胞淤滯綜合徵
較少見,當白細胞計數極度增高時可出現呼吸困難、發紺、臟器梗死、眼底靜脈擴張、視乳頭水腫、眼底出血等表現。
4.其他
胸骨壓痛較常見,進展期時胸骨壓痛明顯,可出現骨痛;細胞破壞、血尿酸升高引起痛風性關節炎;少見有皮膚浸潤,進展期多見。
檢查
 慢性髓系白血病
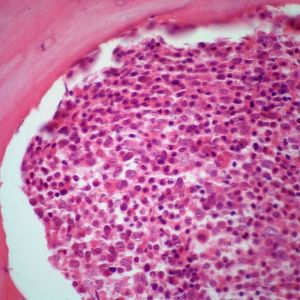
慢性髓系白血病外周血中白細胞升高是主要的特徵,通常高於25×109/L,少數患者在100×10/L以上。確診時紅細胞計數大多正常或輕度變化,隨病情進展呈現輕度貧血,網織紅細胞計數正常或輕度增多。大約50%患者就診時血小板計數高於正常,在慢性期可逐漸升高。外周血原始粒細胞≤5%~10%,加速期外周血原始粒細胞為10%~19%,急變期外周血原始細胞≥20%或者是外周血原始粒細胞+早幼粒細胞≥30%。
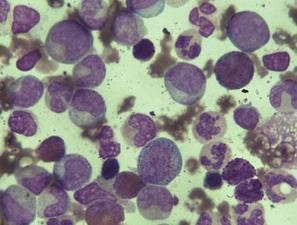
2.骨髓塗片
有核細胞增生極度活躍,以粒系增生為主,紅系增生受抑,粒:紅可達(10~30):1,巨核細胞數量正常或增加,半數患者骨髓區域網路狀纖維增生,部分可發生骨髓纖維化。慢性期骨髓原始粒細胞≤10%,加速期原始粒細胞10%~19%,急變期骨髓原始粒細胞≥20%或者骨髓原始粒細胞+早幼粒細胞≥50%。
3.祖細胞集落培養
慢性期骨髓和外周血粒系、巨核系、嗜酸粒系集落形成增加;進入加速期和急變期後祖細胞增殖和分化能力減弱,集簇增加。
4.中性粒細胞鹼性磷酸酶
90%以上患者成熟中性粒細胞鹼性磷酸酶積分降低或缺失。
5.細胞遺傳學檢測
90%以上的慢粒患者可發現Ph染色體、t(9;22)(q34;q11),是慢粒的標記染色體。
6.分子生物學
使用反轉錄PCR可以檢測出BCR-ABL融合基因。
診斷
根據典型的外周血白細胞計數增高及分類異常、脾大伴有Ph染色體或其變異核型以及BCR/ABL融合基因陽性,即可做出診斷,同時應結合體徵、化驗進行臨床分期。治療
1.慢性期治療(1)藥物治療:甲磺酸伊馬替尼、干擾素、白消安等。
(2)造血幹細胞移植。
(3)白細胞單采。
(4)放射治療。
(5)脾切除。
2.加速期和急變期治療
一旦進入此期應按急性白血病治療,但緩解率低。
