
1疾病簡介
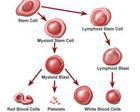 慢性粒單核細胞白血病
慢性粒單核細胞白血病慢性粒單核細胞白血病(Chronic myelomonocytic leukaemia,CMML)是一種慢性髓系白血病。發病率約為1~2/100 000/年,多數在60歲以後發病,男女發病比例約1.5~3:1。具體病因不明。沒有特異性的染色體異位。常見的臨床症狀有發熱、感染、出血、疲乏、體重減輕、盜汗等,約半數病人有肝脾腫大。CMML患者中位生存期約20個月,生存期可能與骨髓原始細胞比例有關。目前尚無特效的治療方法。
2疾病分類
慢性粒單核細胞白血病(Chronic myelomonocytic leukemia,CMML)既往被認為是骨髓增生異常綜合徵(MDS)的一個亞型,因其兼有骨髓發育異常和骨髓增殖的特點,2001年WHO造血和淋巴細胞腫瘤分類將CMML歸屬於骨髓增生異常/骨髓增殖性疾病(MDS/MPD)。
上世紀50年代,Beattie、SINN、Pearson等先後報導“慢性單核細胞白血病”[1~3],1972年,Hurdle等[4]首先報導6例CMML患者,1976年FAB協作組提出急性白血病的形態學分型建議,把MDS另立分型,正式提出CMML包含在MDS之內。2001年WHO分類方案把造血組織和淋巴組織腫瘤按系別分類為髓系腫瘤,淋巴系腫瘤,組織細胞和樹突細胞腫瘤,肥大細胞疾患四大類,CMML被歸入骨髓增生異常/骨髓增殖性疾病(MDS/MPD)。
WHO分類的 MDS/MPD 是一組髓系疾病,兼具即骨髓病態造血和骨髓增殖的雙重特徵。病種包括:慢性粒?單核細胞白血病(CMML)、不典型慢性髓性白血病(aCML)、幼年型粒?單核細胞白血病(JMML)。以往分別歸於MPD或MDS。對CMML是屬於MPD還是MDS一直存在爭議, FAB曾建議將CMML再根據其白細胞計數的高低分為 MDS 樣的 CMML(MDS?like CMML)和MPD樣的CMML(MPD?like CMML),但兩種CMML在生物學行為和預後上並無區別,至今也沒有發現在這兩種CMML中有特徵性的細胞遺傳學和分子生物學差異。臨床上觀察到白細胞低的 CMML,如有脾大,最終都會轉變成“增殖性”並出現明顯的白細胞升高。基於以上原因,WHO新分類不再將CMML分為兩種亞型,而將其統一划歸於MDS/MPD這一新的疾病分類概念中。WHO分類根據骨髓和外周血原始細胞,將CMML再分為CMML?1和CMML?2 兩種亞型。
3分子生物學特徵
MDS/MPD是WHO分型標準中提出的一種新的髓系腫瘤, MDS/MPD?U患者JAK2基因突變率為18.8%(3/16例),CMML患者JAK2基因突變率為12.5%(2/15例),而MDS患者JAK2基因突變檢出率僅為1.4%,提示MDS/MPD?U和CMML患者的發病基礎更與MPD接近。
Ph染色體陽性作為慢性粒細胞白血病的特徵之一,在CMML是陰性的。迄今未發現染色體異位是CMML所特有的。有20%~30%的病人發現基因異常,這些包括染色體數目及結構異常如+8,del(20 q),-7, del(11q),但這些染色體在其他骨髓增殖性疾病及骨髓增生異常綜合徵中也可見到。伴有嗜酸粒細胞增多的病例常表現為t(5;12)(q33;p13),導致TEL基因連線到血小板源性生長因子受體( PDGFbR)基因上,這種情況約見於2%~5%的CMML患者。還有部分患者表現為亨廷頓蛋白(HIP1)基因和前述基因( PDGFbR)融合形成t(5;7)(q33;q11.2)。少數治療相關的CMML也可表現為t(11;16)(q23;p13),從而形成MLL 和 CBP基因融合。也有t(1;13)(p36;q21),t(7;11)(p15;p15) 和 t(8;9)(p11;q34) 等形成的報導。
4臨床表現
發病率約為1~2/100 000/年,男女發病比例約1.5~3:1,多數病人在50歲以上發病,60歲以上發病者約占75%。主要表現為兩組症狀,即與骨髓病態造血相關的症狀及與骨髓增殖相關的症狀。
病人因骨髓增生異常而發生血細胞減少可表現為乏力、心悸、蒼白、低熱、感染或出血等。表現在骨髓增殖性上的特點如:異常單核細胞增生,且這種細胞有浸潤的特徵,如皮膚、腺體、齒齦、骨等髓外浸潤,淋巴結、肝脾腫大,甚至巨脾。有作者報導[5]24例患者中,50%病人出現肝大,45.8%出現脾腫大,20.8%出現淋巴結受累。
根據CMML的特點,可以將CMML分為慢性期、加速期、急性變期。慢性期患者病情相對穩定,無症狀或僅有低熱、乏力等,少見肝脾淋巴結腫大。進入加速期後病情進展迅速,尤其是單核細胞明顯增多者,病情極重,多在急性變之前即死於出血、感染或臟器衰竭。也有缺乏加速期而從慢性期轉為急性變者。
5臨床診斷
1實驗室檢查
 慢性粒單核細胞白血病
慢性粒單核細胞白血病血常規檢查具有多樣性。常常表現為貧血和白細胞增高,主要是粒細胞和單核細胞增高,外周血單核細胞絕對數>10×109/L,可出現幼稚單核細胞。粒細胞常出現成熟粒細胞增多,有或無粒系發育異常,可見未成熟的粒細胞。部分病人也可以出現粒細胞減少。多數患者血小板降低,但也有部分病人血小板計數正常或增高。骨髓穿刺檢查,增生程度相差較大,多數為明顯活躍或極度活躍,也有增生低下者。粒細胞和單核細胞增多。原始粒細胞多>5%,原幼單核細胞也>5%,但二者之和<20%。紅系及巨核系增生減低。三系均有不同程度的病態造血,其中紅系的典型表現為巨幼樣變、核碎裂、花瓣樣核、多核紅細胞,成熟紅細胞嗜鹼性點彩。粒系病態造血表現為顆粒減少、核分葉過少(P?H畸形)、胞漿中出現空泡等,顆粒過多作為一種病態表現,特異性較差。巨核系典型的病態造血為小巨核細胞、淋巴樣巨核細胞、單圓核巨核細胞、多圓核巨核細胞。
血漿和尿溶菌酶含量是幾乎總高的。 這種溶菌酶是由單核細胞分泌的,並與單核細胞在體內的數量成正比。
2表現診斷
WHO對CMML的診斷標準較FAB的未作大的變動。(1)持續性外周血單核細胞增多>1×109/L;(2)Ph(-),BCR/ABL(-);(3)外周血和骨髓中原始細胞<20%;(4)髓系中1個或1個以上細胞系別有發育異常,如果無發育異常或極微,但其他條件符合,且有以下表現者,仍可診斷為CMML:骨髓細胞有獲得性細胞遺傳學克隆異常,或單核細胞增多已持續3個月以上,而且排除所有能引起單核細胞增多的原因;(5)外周血中原始細胞<5%,和BM中<10%者,診斷為CMML?1。外周血原始細胞5%~19%或骨髓10%~19%,或外周血或骨髓原始細胞<20%而Auer小體陽性,診斷為CMML?2;(6)符合以上診斷標準且外周血嗜酸粒細胞≥1.5 ×109/L者,診斷為CMML?1或CMML?2伴有嗜酸粒細胞增多。(診斷CMML時,原始細胞包括原始粒細胞、原始和幼稚單核細胞)。轉貼於 中國論文下載中心
3鑑別診斷
6.1 骨髓增生異常綜合徵(MDS) CMML曾作為MDS的分類之一,兩者臨床表現及實驗室結果相似,但CMML的外周血單核細胞增多>1×109/L。
6.2 慢性粒細胞白血病(CML) CML也是骨髓增殖性疾病之一。具有骨髓增殖性疾病的特點,骨髓增生程度多為活躍至極度活躍,以粒係為主,其他系均受抑制。具有特異性的Ph染色體陽性。
6.3 不典型慢性髓性白血病(aCML) 所謂不典型CML是指Ph(-),又無bcr?abl易位的CML。外周血白細胞正常或減少,血片內幼稚粒細胞增多(>0.15),但無嗜鹼粒增多。骨髓示增生活躍或極度活躍、伴多系病態造血,以粒系增生為主,粒:紅之比常<10:1,嗜酸粒細胞正常或增多,但嗜鹼粒不增多。已知本型用酪氨酸激酶抑制劑(STI571,glivec,格列衛)效果差。
6.4 幼年型粒?單核細胞白血病(JMML) 本型是一種罕見的嬰幼兒期,多見於2歲以下的嬰幼兒,兼有MDS/MPD雙重特性的混合型髓細胞系腫瘤。患兒常伴皮膚表現(如濕疹和黃瘤病等)。JMML以顯著的髓系細胞病態造血、單核細胞增多、外加抗鹼血紅蛋白(HbF)升高為特徵。患兒Ph(-),也無bcr?abl重排,髓內幼稚和異常單核細胞,以及紅系前體細胞增生、紅系細胞生成異常表現在HbF升高(0.15~0.50),以及紅細胞I抗原表達降低。
6治療及預後
所有專家都同意原始細胞比例與預後密切相關,原始細胞越高預後越差。此種疾病的存活時間10~60個月,中位生存期20個月。到目前為止,沒有更好的治療方法可以使病人獲得緩解,對於新診斷的CMML病人,探討更有意義的實驗性治療方案是需要我們大家去努力。
7.1 支持治療 在目前沒有更好治療方法的情況下,支持治療是CMML的關鍵性治療。多數CMML患者年齡較大,並常伴有其他疾病,支持治療尤為重要。血紅蛋白 <10 g/L 的患者,根據貧血症狀,定期輸注濃縮紅細胞。血小板<20×109/L伴出血症狀時,進行血小板輸注,血小板<10×109/L 時,進行預防性血小板輸注。其他抗感染、營養支持等均要選擇性套用。
7.2 化療 CMML具有明顯的病態造血,造血功能多數較差。多數病人不能耐受強烈化療,而小劑量化療或許能起到一定療效。常用的單藥治療方案有:小劑量阿糖胞苷、5?氮雜胞苷、高三尖杉酯鹼、依託泊苷等均有一定的作用。
高白細胞患者可用羥基脲。羥基脲用量為1~4 g/d,使白細胞維持在(5~10)×109/L 水平。
 慢性粒單核細胞白血病
慢性粒單核細胞白血病有報導套用拓撲異構酶Ⅰ抑制劑拓撲替康(topotecan)治療CMML取得一定療效。臨床試驗,25例CMML患者套用topotecan,2 mg/(m2·d),持續靜脈滴注5 d,28%的患者獲完全緩解(CR),中位緩解期為8個月[6]。7.3 去甲基化的治療 脫甲基藥物如地西他賓(decitabine,5?氮雜胞嘧啶脫氧胞苷,5?氮雜?2’?脫氧胞苷)為目前已知最強的DNA甲基化特異性抑制劑,在體內能使因甲基化而失活的抑癌基因再轉錄,誘導其表達。與加強化療相比,通常這些藥物不會出現嚴重的不良反應。Aribi報導[7],用去甲基化藥物地西他賓治療19例CMML患者,11例完全緩解,2例部分緩解,總有效率69%。並且沒有出現嚴重併發症者。Wijermans等[8]分析了3個臨床試驗的結果,總數為31例的CMML病人參與治療,平均年齡70.2歲,71%為男性患者,就診時有8/28 (29%)病例白細胞數>2 000/L,骨髓原始細胞比例>5%的占39%(11/28)。總有效率25% (14% CR+11% PR),血液學進步者有11%,病情穩定者占39%。
7.4 造血幹細胞移植 可用於有相合供者的<50歲的患者。造血幹細胞移植理論上是有可能治癒CMML的方法。但年齡等條件限制使能夠移植治療的病人數較少。
7.5 其他臨床試驗性治療 脫乙醯組蛋白的抑制劑如丙戊酸鈉顯示出令人滿意的療效,Siitonen等[9]報導套用丙戊酸、維A酸、1,25二羥維生素D3治療19例MDS或CMML患者,3例有效,9例因副作用在16周內停止治療。
反應停100~200 mg/d,口服,約20%的患者減少輸血量或提高血紅蛋白。
在某些病人中,第5和12染色體發生易位,導致血小板源性生長因子受體基因beta (PDGFR?b) 與TEL基因出現。這種基因表達的蛋白質可以被酪氨酸激酶抑制劑如格列衛抑制。有人報導1例CMML病人伴PDGFR?b?TEL基因易位在進行造血幹細胞移植後復發,套用格列衛有效。提示患有CMML的部分病人可以用這種藥物進行成功治療。
但就目前的醫療技術來看,恐怕只有骨髓移植才能完全治癒該病,這也是我們需要努力去改變的現狀。
