流行病學
以青壯年男性發病率最高目前沒有其他相關內容描述。
病因
本病病因不明有認為與常染色體顯性遺傳、自身免疫異常和纖溶活性降低及慢病毒感染等有關。
發病機制
發病機制還不清楚。有學者認為與常染色體顯性遺傳、自身免疫異常和纖溶活性降低及慢病毒感染等有關。
組織病理:主要病變為真皮下部和皮下脂肪組織內的細小動脈內膜炎。內皮細胞腫脹和增生,由於PAS陽性物質沉積而管壁增厚,血栓形成,以致產生缺血性梗死,發生楔形潰瘍。中膜和外膜一般不受累,內彈力膜正常缺血區內膠原纖維腫脹、變性或漸進性壞死,最後完全纖維化而萎縮早期病灶內可有黏蛋白沉積,至晚期則只見於周圍組織內。可見小腸漿膜下散在分布的梗死性潰瘍、白色瘢痕或穿孔。其組織病理變化與皮膚損害相同。
臨床表現
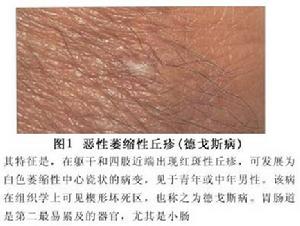 惡性萎縮性丘疹病
惡性萎縮性丘疹病以青壯年男性發病率最高,通常是累及皮膚和腸道,而皮膚往往在先,約1/3病例只有皮膚損害。約20%病例累及中樞神經系統,少見的尚可累及眼心、腎和膀胱等皮膚損害主要分布於軀幹和四肢,特別是在背部和肢體近端,而面和手足較少。原發損害為直徑2~15mm的半球狀水腫性紅色丘疹病程中部分損害持續存在或吸收消失,遺留小的白色皮膚萎縮,而大多數中央迅速壞死,發生潰瘍,遺留瓷白的皮膚萎縮斑,上附有灰白色鱗屑,有狹窄的高起邊緣其上或有擴張的毛細血管。損害成批出現,少則幾個多則百餘個散在分布較少相互融合。一般無自覺症狀可持續數個月或數年。在皮膚損害出現後3周~10餘年間發生腸損害多數是小腸、大腸、腸系膜,而胃亦有累及呈隱襲發病早期可僅為消化不良腹瀉或便秘等,進而發生腹絞痛和便血,最後因發展為多發性腸穿孔和腹膜炎故預後不良。剖腹探查,可在腸壁上發現多發性卵圓形針尖至直徑為2cm的漿膜下白色萎縮性瘢痕。無腸損害的患者中,有33%~50%並不產生嚴重後果。眼部損害為在眼瞼、球結膜可有白色無血管斑,脈絡膜和視網膜缺血變性復視、視盤水腫和視神經萎縮等。
併發症:
腸損害後可發生腹絞痛和便血,最後因發展為多發性腸穿孔和腹膜炎。眼部損害球結膜可有白色無血管斑,脈絡膜和視網膜缺血變性、復視、視盤水腫和視神經萎縮等。
診斷
根據臨床表現和組織病理檢查可以確診。
鑑別診斷:
但有時需與淋巴瘤樣丘疹病、急性痘瘡樣苔蘚樣糠疹和變應性皮膚血管炎相鑑別。
檢查
實驗室檢查:
免疫球蛋白尤以IgA顯著升高,纖維蛋白原含量升高和血小板凝集試驗為陽性。此外一般無陽性發現。
其它輔助檢查:
組織活檢,主要病變為真皮下部和皮下脂肪組織內的細小動脈內膜炎
治療
主要是對症治療。一般可用吲哚美辛(消炎痛)、阿司匹林與雙嘧達莫(潘生丁)。雖可用皮質激素但在晚期應注意發生腸穿孔的可能。亦可試用肝素治療。
預後
在皮膚損害出現後3周~10餘年間發生腸損害,多數是小腸、大腸、腸系膜,而胃亦有累及。呈隱襲發病,早期可僅為消化不良、腹瀉或便秘等,進而發生腹絞痛和便血,最後因發展為多發性腸穿孔和腹膜炎,故預後不良。
預防
加強護理和營養:以提高患者的抵抗力和免疫力。預防感染 應注意隔離,儘量減少與病原體的接觸。避免近親結婚,進行婚前檢查生育諮詢。
