疾病概述
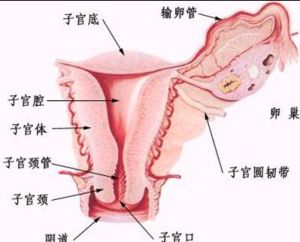 子宮
子宮先天性卵巢發育不全,為女性缺少一條X染色體所致的身材矮小、原發性閉經、頸蹼、肘外翻等異常。本型發病率遠比上一型低,約占女性智力缺陷的0.64%,其臨床特點為患者外貌女性,身體較矮,第二特徵發育不良,卵巢缺如,無生育能力。部分患者智力輕度低下。有的患者伴有心、腎、骨骼等先天畸形。
症狀體徵:
1、身材矮小為本病最恆定的特徵。
2、智力低下。
3、本病患者通常顯幼稚、溫順,容易相處。
4、患者外生殖器呈幼女型、性腺不發育,子宮及輸卵管小,卵巢呈條索狀,卵母細胞和囊狀卵泡常缺如,原發性閉經、不育,陰毛稀少、陰道黏膜薄,無分泌物。
5、可有眼鹼下垂、內眥贅皮、後髮際低、低位大耳、高齶弓、頸蹼、黑色素痣等。常並有骨胳畸形。
病因
1959年Ford等證實該病因性染色體X呈單體性所致。Turner綜合徵的表型是女性在活產女嬰中約占0.4‰,其發生率低是因為X單體的胚胎不易存活,約99%的病例發生流產。該病也是人類唯一能生存的單體綜合徵。
病理生理:患者的性腺發育障礙,卵巢被條索狀纖維組織所取代。
診斷檢查
患兒血清雌二醇水平低,濾泡刺激激素(FSH)、黃體生成素(LH)明顯增高。性染色質檢查為陰性。確診必需作染色體檢查,其核型有以下幾種類型:
①單體型:45,X0,是最多見的一型,具有典型症狀。
②嵌合型:45,X0/46,XX,若以46,XX細胞為主,症狀多數較輕,約20%可有青春期發育,月經來潮,部分可有生育能力,但其自然流產率和死胎率均高、且子代患染色體畸變的風險率亦高。
③X染色體結構畸變型:一條X染色體長臂和或者短臂缺失,如46,Xdel(Xq)或46,Xdel(Xp),還有X等染色體,如46,Xi(Xq)或46,Xi(Xp)。
治療方案
改善其成人期最終身高和性徵發育,保證患兒心理健康。爭取早期確診,儘早使用基因重足人生長激素,每晚0.15U/kg皮下注射,可使患兒身高明顯增長。若其骨齡落後明顯,可合併使用司坦唑醇(康力龍)每日25—50μg/kg口服,效果更好。同時定期檢測甲狀腺功能和骨齡發育情況,當骨齡達12以上時,可開始給予口服小劑量雌激素治療,以促進乳房和外生殖器發育。常用的有炔雌醇(10—20μg/d)或己醇雌酚(0.1—0.5mg/d)或妊馬雌酮,從每日310μg開始,根據臨床效果逐步加量。
安全提示:
1、本疾病患者不可能建立生育功能。為了第二性徵的發育和維持,可長期套用雌激素替代療法,同時可防止骨質疏鬆。但長期套用任何類的雌激素,都有誘發子宮內膜癌的危險。
2、主張用雌、孕激素人工周期療法,以誘發周期性子宮出血,一方面對患者是一種心理安慰,另一方面由於子宮內膜定期剝落,可預防子宮內膜癌的發生。
3、對身材矮小者,採用苯丙酸諾龍治療,效果良好。但骨骺已癒合者,療效差。對嵌合體含Y染色體者,應切除雙側性腺,以防惡變。
