原理
樣品經消化後,在pH4.0~4.5時,鋅離子與二硫腙生成紫紅色配合物,溶於四氯化碳,與標準系列相比較進行定量。
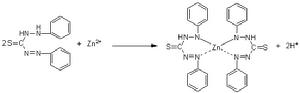 反應方程式
反應方程式試劑
1.C(CH3COONa)=2mol/L乙酸鈉溶液:稱取68g乙酸鈉(CH3COONa·3H2O),加水稀釋至250ml。
2.C(CH3COOH)=2mol/L乙酸溶液:量取10.0ml冰乙酸,加水稀釋至85ml。
3.乙酸-乙酸鹽緩沖溶液C(CH3COONa)=2mol/L乙酸鈉溶液與C(CH3COOH)=2mol/L乙酸溶液等體積混合,此溶液PH為4.7左右。用0.01%二硫腙四氯化碳溶液提取數次,每次10ml,除去其中的鋅,至四氯化碳層綠色不變為止,棄去四氯化碳層,再用四氯化碳提取乙酸-乙酸鹽緩衝液中過剩的二硫腙,至四氯化碳層無色,棄去四氯化碳層。
4.1:1氨水。
5.C(HCl)=2mol/L鹽酸:最取10ml鹽酸,加水稀釋至60ml。
6.C(HCl)=0.02mol/L鹽酸:吸取1ml C(HCl)=2mol/L鹽酸,加水稀釋至100ml。
7.酚紅指示劑:0.1%乙醇溶液。
8.20%鹽酸羥胺溶液:稱取20g鹽酸羥胺,加60ml水,滴加1:1氨水,調節PH至4.0-5.5,以下操作同乙酸-乙酸鹽緩衝溶液的處理。然後加水稀釋至100ml。
9.25%硫代硫酸鈉溶液:用C(CH3COOH)=2mol/L乙酸溶液調節PH至4.0-5.5。以下按試劑3中用0.01%二硫腙-四氯化碳溶液,處理。
10.0.01%二硫腙-四氯化碳溶液。
12.鋅標準溶液:準確稱取0.1000g鋅,加10mlC(HCl)=2mol/L鹽酸,溶解後移入1000ml溶量瓶中,加水稀釋至刻度。此溶液每毫升相當於100μg。
13.鋅標準使用液:吸取1.0ml鋅標準溶液,置於100ml的容量瓶中,加1mlC(HCl)=2mol/L鹽酸,以水稀釋至刻度。此溶液每毫升相當於1μg鋅。
儀器
分液漏斗、分光光度計。
操作方法
1.樣品的消化:同“砷的測定”中硝酸-高氯酸-硫酸法或硝酸-硫酸法。
2.標準曲線的繪製
吸取0.0、1.0、2.0、3.0、4.0、5.0ml鋅標準使用液,分別置於125ml分液漏斗中,各加入C(HCl)=0.02mol/L鹽酸至20ml。漏斗中,備加c(HcL)=o.02M1兒鹽酸至20m1。然後分別加入10ml乙酸-乙酸鹽緩衝液、1m125%硫代硫酸鈉溶液,搖勻,再各加入10.0ml二硫腙使用液,劇烈振搖2min。靜置分層後,四氯化碳層經脫脂棉濾入lcm比色杯中,以零管調節零點,于波長530nm處測定吸光度、繪製標準曲線或求出回歸方程。
3.樣品溶液和試劑空白液油定
吸取5.0ml~10.0ml消化後定容的樣品溶液和相同量的試劑空白液,分別置於125m1分液漏斗中,加5m1水、0.5ml20%鹽酸羥胺溶液,搖勻,再加2滴酚紅指示劑,用1:1氨水調至紅色,再多加2滴。再加5ml 0.01%二硫腙-四氯化碳溶液,劇烈振搖2min,靜置分層。將四氯化碳層移入另一分液漏斗中,水層用少量二硫腙-四氯化碳溶液振搖提取,每次2mL~3ml,直至二硫腙-四氯化碳溶液綠色不變為止。合併提取液,用5m1水洗滌,四氯化碳層用C(HCl)=0.02mol/L鹽酸提取2次,每次10ml,提取時劇烈振搖2min,合併C(HCl)=0.02mol/L鹽酸提取液,並用少量四氯化碳洗去殘留的二硫腙。以下自“加入l0ml乙酸-乙酸鹽緩衝溶液”起,按標準曲線繪製操作方法進行操作。根據測得吸光度從標準曲線上查得相當於鋅的含量,或將吸光度代入回歸方程求得鋅的含量。
計算
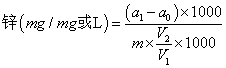
‍式中:
a1——測定用樣品消化液中鋅的含量(μg)
a0——試劑空白液中鋅的含量(μg);
m——樣品質量(體積)[g(ml)];
V1——樣品消化波速體積(nII);
V2——測定用樣品消化液的體積(m1)。
注意事項
1.測定時加入硫代硫酸鈉、鹽酸羥胺和在控制pH值的條件下,可防止銅、汞、鉛、鉍、銀和鎘等離子的干擾。並能防止二硫腙被氧化。
2.所用玻璃儀器用10%一20%硝酸浸泡24h以上,然後用不含鋅的蒸餾水沖洗潔淨。
3.硫代硫酸鈉是較強的配合劑,它不僅配合干擾金屬,同時也與鋅配合.所以只有使鋅從配合物中釋放出來,才能被二硫腙提取,而鋅的釋放又比較緩慢,因此必須劇烈根據2min。
