
丙酮酸脫氫酶複合物
 丙酮酸脫氫酶複合物
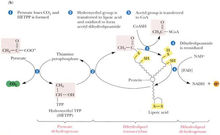
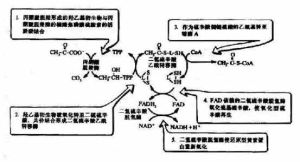
丙酮酸脫氫酶複合物PDHC 由6 種酶組成,其中丙酮酸脫羧酶(pyruvate decarboxynase, E1, EC 1.2.4.1)、二氫硫辛酸轉乙醯化酶(dihydrolipoamide transacetylase, E2,EC 2.3.1.12)和二氫硫辛酸脫氫酶(dihydrolipoamide dehydrogenase, E3, EC 1.8.1.4)是其3種主要的酶。PDHC核心由60個E2亞單位組成,連線30個拷貝的E1分子和6個分子的同型二聚體E3 蛋白。E1亞單位是由2個α亞單位和2個β亞單位組成的異構四聚體。PDHC還包括2個調節亞單位, 即:E1-激酶(PDK)和E1-磷酸酶(PDP)。PDK 通過催化E1亞單位上的3 個絲氨酸位點的磷酸化而使複合物失活, 在哺乳動物組已經識別出該酶的4種異構體形式。PDK 在不同的組織分布不同,PDK1主要在心臟表達,PDK2則廣泛表達,PDK3在睪丸表達,PDK4在骨骼肌和心肌表達。PDP存在2種不同的異構形式,PDP1優先在肌肉組織表達,而PDP2在肝臟組織表達。且PDP2與PDP1不同,不需要Ca2+的調節。PDHC還含有一個稱為蛋白X( protein X) 的成分,現已證明是二氫硫辛酸脫氫酶結合蛋白即E3結合蛋白( E3 binding protein, E3BP) 。E2核心和E3的結合是通過12個分子的E3BP連線起來的。它主要存在於人的骨骼肌和心肌組織,在其他組織只有少量的表達。它可能在複合物的組裝過程中發揮作用。此外,PDHC還需要3種輔酶:硫辛酸焦磷酸鹽、硫辛酸和黃素腺嘌呤二核苷酸。
PDHCDNA和cDNA
丙酮酸脫羧酶E1α亞單位(PDHA1)的基因組DNA 全長15.92 kb,含有11 個外顯子。位於X 染色體短臂上(Xp22.1- 22.2)。其中含有保守的硫辛酸焦磷酸鹽(TPP)結合區,它位於外顯子6編碼195胺基酸殘基和外顯子7 的255胺基酸殘基之間。此外,在4號染色體上有一段與PDHA1同源的無內含子的序列,主要在睪丸
 丙酮酸脫氫酶複合物
丙酮酸脫氫酶複合物丙酮酸脫氫酶複合物缺陷與疾病
臨床表現
PDHC 缺陷是導致線粒體能量代謝障礙最常見的原因之一,同時也是兒童乳酸酸中毒和早發性退行性神經變性病的最常見病因。腦內乙醯輔酶A幾乎都來源於丙酮酸,所以PDHC的缺乏常導致多種神經系統損害。按照Robinson等提出的標準,患者的臨床表現可分為三級,Ⅰ級:病人出生後早期即患有嚴重的乳酸血症,PDHC 活性極低,男性患兒多於胚胎時期發病,導致流產、死胎、先天性紋狀體發育不全、缺氧缺血性腦病,常於新生兒早期死於乳酸酸中毒。Ⅱ級:乳酸血症較Ⅰ級輕,出生時正常,智力運動及體格發育落後,患兒多於嬰兒期死亡,少數存活到十幾歲。Ⅲ級:患者乳酸血症較輕,PDHC殘存活性多高於20%。E3BP 缺乏患者在神經病理檢查中最常見的表現是Leigh 綜合徵,胼胝體變薄或缺失、基底節對稱性壞死性病變;同時E3BP缺乏的病人PDHC 酶的殘存活性相對較高。
PDHA1缺乏症的臨床表現PDHA1缺陷是丙酮酸脫氫酶缺乏症最常見的原因。由於PDHA1的絲氨酸位點對於整個PDHC酶活性的調節都有重要的作用,所以發生於這個亞單位的突變最多。E1α亞單位缺乏的臨床表現在不同的年齡段有一定的差異。在新生兒期患者主要表現為乳酸酸中毒和腦發育不全。嚴重的新生兒期表現常提示重要的蛋白調節區的異常。一些嬰兒期起病的患者在5 歲之前發展成為Leigh 綜合徵,表現為發育遲緩、驚厥、間歇性無力、共濟失調、腦性癱瘓等進行性神經系統病變,並伴有基底節和腦幹損害。少數患者起病時較輕,在給予高碳水化合物飲食後出現間歇性的短暫的共濟失調,緩慢進展,數年後逐漸發展為Leigh綜合徵。
PDHA1缺陷為X 連鎖遺傳性疾病,患者總體男女數量基本相等,但是臨床表現差異顯著,考慮、與生化缺陷的嚴重性和基因突變的特異位點有關。男性發病更早更重,表現為胎兒期死亡、新生兒乳酸酸中毒、Leigh綜合徵和間歇性共濟失調。女性患者的臨床表現更為複雜,多發畸形較常見, 如伴有前額腫塊的小頭畸形、寬鼻橋、鼻上翻、長人中和鼻翼搧動等。還可見到耳低位, 前置肛門,手指、上臂短。合併乳酸酸中毒的女性患者預後不良。女性雜合子臨床表現複雜,嚴重患者於胎兒期~嬰兒期死亡,輕型患者終身不發病, 一些嚴重酶缺失的女性患者仍然能夠存活,主要決定於X 染色體隨機失活的形式。
PDH E1β亞單位缺乏的臨床表現Brown 等首先在2個病人中發現E1β亞單位缺乏。該基因缺陷的主要表現為乳酸酸中毒和肌張力低下。
E2亞單位缺乏的臨床表現E2缺乏的患者未見有神經系統的改變,主要見於患有膽汁性肝硬化的患者。這些患者多有抗二氫硫辛酸轉乙醯化酶的自身抗體。
E3和E3結合蛋白缺陷的臨床表現E3和E3結合蛋白的缺陷很少見。所報導的患者父母多為近親婚配,屬常染色體隱性遺傳。在三羧酸循環和支鏈胺基酸代謝中E3也參與其他2個脫氫酶的組成。E3結合蛋白缺乏的男性患者臨床表現與、PDHA1 缺陷的男性患者相似, 主要表現為體格、智力運動發育落後,肌張力低下、乳酸酸中毒和Leigh綜合徵。對於乳酸酸中毒合併α-酮酸尿症和血漿支鏈胺基酸水平增高的患者應高度懷疑E3缺乏。
PDHC基因的突變
關於PDHA1 基因的研究較為深入,迄今已發現82 種突變,其中大部分為無義或錯義突變,為43個,除外顯子2 外均發現過突變,其中以外顯子3、7、8、11 最為多見。而無義或錯義突變多見於外顯子3、7 和8,缺失和插入突變主要見於外顯子10 和11。絕大多數男性患者攜帶無義或錯義突變,相反女性則多攜帶缺失或插入突變。Naito等報導的硫辛酸治療有效的患者中PDHA1基因突變包括以下位點:H44R、R88S、G89S、R263G、V389fs、V71A 和C101F,其中H44R、V71A、R88S 和G89S位於外顯子3 上,提示PDHA1基因外顯子3 突變患者對硫辛酸治療反應較好。11例患者PDHA1基因外顯子8存在R263G突變,是最多見的突變形式。
關於PDHC其他基因的研究較少,2004年Brown等在2例無關聯患者中發現了E1β亞單位2種不同的突變Y132C和P344S。E2亞單位的突變極為少見。迄今已報導的E3 亞單位的突變有9個,其中7個為錯義或無義突變,E3結合蛋白基因突變6個。
診斷
患者出現智力運動落後、無力、肌張力低下等可疑PDHC 缺陷臨床表現時,可進行以下檢查。
血液中丙酮酸脫氫酶複合物酶活力下降,酶活力檢測是診斷該病的金標準。
血、腦脊液乳酸和丙酮酸水平的測定:患者血清乳酸和丙酮酸水平常顯著升高,而乳酸/丙酮酸比例正常。如果血清乳酸正常,需要測定腦脊液乳酸和丙酮酸水平,患者腦脊液乳酸和丙酮酸水平通常升高,乳酸/丙酮酸比例正常。
丙酮酸脫羧酶活性測定:測定皮膚成纖維細胞、成淋巴細胞、肌肉組織或腦組織酶活性。
檢測PDHE1α亞單位、E1β亞單位、E3和E3結合蛋白基因是否存在突變。
影像學檢查:Leigh綜合徵患者可見腦幹和基底節對稱性病變,部分患者表現為腦發育不全、腦室擴張、皮質萎縮、胼胝體發育不全、側腦室移位等異常。
神經病理學的檢查:患者胼胝體可有不同程度缺失,也可伴有其他缺陷,如:小腦普肯野氏細胞和齒狀核不發育等異常。Leigh 綜合徵患者基底節、被蓋部灰質、腦幹、小腦、脊髓可見對稱性局灶性壞死、神經元消失、脫髓鞘改變和血管增生壞死。
治療與預後
對於線粒體疾病的治療,還沒有令人滿意的方法。對於PDHC 缺陷患者,生酮飲食、硫辛酸、二氯醋酸、左鏇肉鹼、輔酶Q10 有一定療效。Brown等認為對於乳酸酸中毒較輕、發育正常的男性PDHC 缺乏患者,生酮飲食的治療效果最好。如果患者對硫辛酸有反應,可以在生酮飲食治療的基礎上輔以TPP治療,可能會產生較好的治療效果。二氯醋酸被認為是最有潛力降低乳酸水平的藥物,能刺激糖的有氧氧化限速步驟。但是,一些學者認為儘管二氯醋酸可以通過對正常細胞酶的激活使突變細胞釋放的乳酸減少,對於提高突變細胞的丙酮酸氧化意義不大。
PDHA1亞單位基因缺陷患者中對硫辛酸反應的患者預後較好,研究表明患者體內硫辛酸的濃度與其神經系統症狀的改善呈正相關,而對於硫辛酸無反應的患者預後不良。對於基因診斷明確的患者基因治療將是值得期待的方法。
