X射線吸收精細結構(XAFS)譜是用於描繪局部結構最強有力的工具之一。在此技術中,我
們將X射線能量調整至與所研究的元素中內電子層一致,再用於探測樣品,然後監測吸收的X射
線數量與其能量的函式關係。如果採用足夠的精確度,光譜會展現出小的振盪,那是局部環境對
目標元素基本吸收機率影響的結果。從光譜中,我們還能得到吸收原子與鄰近原子的間距、這些
原子的數量和類型以及吸收元素的氧化狀態,這些都是確定局部結構的參數。通過選擇不同能量
的X射線,我們可以獲得樣品中所有元素的此類信息。
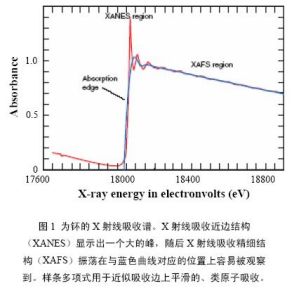 XAFS
XAFS圖1為一個X射線區域吸收譜的例子。當X射線的能量與樣品中某一元素的一個內電子殼層的能量發生共振時,會出現突然的升高電子被激發形成連續光譜(圖1為鈽的X射線吸收譜)。由於光譜的形狀,該光譜又被稱為吸收邊。多數情況下,吸收邊分得很開,且目標元素只是通過掃描一個合適的能量範圍來簡單的選擇。沿著吸收邊,隨著X射線能量的增加,當X射線的穿透深度變大時,吸收率單調下降。當光譜被擴展越過一個特定邊緣時,可觀察到精細結構。當超過20到30電子伏特寬的譜峰和譜肩剛通過邊沿的起點時,就出現了X射線吸收近邊結構(XANES)區。位於能量衰減至幾百個電子伏特的邊沿的高能量一側的精細結構被稱為X射線吸收精細結構(XAFS)。XANES和XAFS中的精細結構已被研究得較透徹,它使XAFS能用於確定化學物質的種類與局部結構。在邊區以外,XAFS精細結構以一系列起伏振盪的形式疊加在本應為孤立原子所具有的較為平滑的吸收曲線上。這些精細結構是由於電離出的光電子波與鄰近原子對部分這些波的反向散射波之間干涉而形成的。隨著X射線能量的改變,干涉條件也發生相應改變,致使鄰近原子產生了振盪式的精細結構。
XAFS的調節可通過一個單散
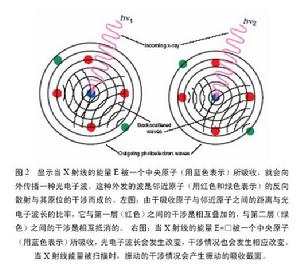 XAFS
XAFS射公式來描述,對化學家而言,公式
中包含一些重要的結構測量參數。這
些參數(並非所有的)包括與吸收原
子距離相等的相同原子序數的相鄰
原子數(即鄰近原子的電子層),一
個與Z相關的該電子層中每個原子
的反向散射振幅函式,對偶的德拜-
沃勒(Deby-waller)因子,以及一個
獨特的吸收體-散射體對的相移特
征。
在數學上,可以對所有電子層的
散射進行累計,從而得出合成的
XAFS光譜。這些測量參數從非線形
最小二乘法曲線擬合的數據中被提
取出來。首先設計出來的模型由許多原子的相鄰電子層(必要時可通過多散射路徑加以補充)組
成。XAFS是由單散射方程計算出的這些電子層獨立波的總和。
擬合必需的相和振幅源從結構類似於標準組分的、有序的光譜,變成了任意排列的、與最終
結構十分接近的原子簇非常精確的從頭開始的計算。
對於不同的元素,相移和振幅各不相同,隨著Z增加,能夠確認出元素類型的相移和振幅的
差異大體上為±3~4。結構參數,例如R,N,有時是σ,允許上下浮動,直到數據和擬合曲線
之間最小二乘差達到最小。附加的化學信息,如在不同參數或電子層之間的相關性,或一個參數
的容許範圍都作為約束條件而被引入。
把XAFS當作一種正弦波的疊加的觀點需要用傅立葉分析,通過傅立葉變換從χ(k)轉變到χ
(R)。由於該變換把每一個波都變成一個峰,χ(R)與吸收體周圍的數目加權平均徑向結構函式相
關,而且模量確實意味著在吸收體周圍存在成對的分布。由於這個原因,儘管大多數分析實際上
發生在K空間(χ(k)),傅立葉變換表達式(χ(R))——通常只是模量——也經常用於圖表中。
數據經常表現為作為加權光譜(χ3(k)),以強調較長距離造成的散射作用。
姜美花譯自《錒系元素季刊》2004,2
胡鳴怡校
