
測序目的
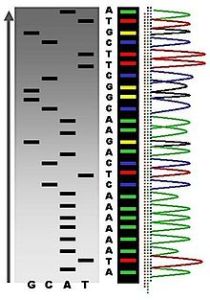 自動化chain-termination DNA測序結果的一個例子
自動化chain-termination DNA測序結果的一個例子1、測定未知序列
2、確定重組DNA的方向與結構
3、對突變進行定位和鑑定
4、比較研究
發展歷史
70年代末,WalterGilbert發明化學法、FrederickSanger發明雙脫氧終止法手動測序,同位素標記;
80年代中期,出現自動測序儀(套用雙脫氧終止法原理)、螢光代替同位素,計算機圖象識別;
90年代中期,測序儀重大改進、集束化的毛細管電泳代替凝膠電泳;
2001年完成人類基因組框架圖。
國內現狀
人類基因組計畫、基因晶片、個性化分子診斷、生物雲計算……這些在21世紀第一個十年里吸引無數眼球的熱門辭彙,都和一個產業頗有淵源——DNA測序。生物技術和信息技術在這片創意新天地里水乳交融,如果用一句詩來形容坐擁兩大技術護航的DNA測序產業,那就是——天生麗質難自棄。
在業內人士眼裡,DNA測序出身高貴,它破解基因密碼(即鹼基序列),將基因組學與IT技術相結合,發展出一門新興學科——生物信息學。以它為代表的基因技術,顛覆了傳統生物學技術,引領生命科學未來發展潮流。以它為代表的基因工程,在醫療健康、環境保護、新能源、新材料、現代農業等熱門領域大顯身手。
在業內人士眼裡,DNA測序足夠高科技,堪稱“一項新技術衍生出一個新行業”的典範,在短時間內迅速成為國內外VC和PE的寵兒,發展速度之快以至於沒有人能準確描繪出它十年後的發展藍圖。在日新月異的DNA測序技術面前,任何預測可能都顯得保守。
高科技領域就是這樣一個誕生傳奇的地方。據《全球DNA測序行業商業模式與投資預測分析報告前瞻》調查,DNA測序已從一項令人高山仰止的前沿技術迅速普及為生命科學常規技術。DNA測序成本下降的速度幾乎可與電腦晶片運算能力增強的速度匹敵——過去一個微生物全基因組DNA測序需要花費300-500萬元,而現在它的成本只有30-50萬元。DNA測序的發展不僅體現在成本的降低,更表現在高通量測序使得工作效率得到了大幅提高,這就為DNA測序產業化鋪平了道路。
在DNA測序商業化的浪潮下,我國《生物產業發展“十二五”規劃》提出完成10000種微生物、100種動植物基因組測序、發現約500個新的功能基因、轉化套用5個以上有重大經濟價值的基因或蛋白。按照每種微生物進行“基因組完成圖”測序的費用為30-50萬元來看,DNA測序帶來的市場容量達千億元,這還僅僅是DNA測序商業套用市場的冰山一角。
測序原理
化學修飾法
化學試劑處理末段DNA片段,造成鹼基的特異性切割,產生一組具有各種不同長度的DNA鏈的反應混合物,經凝膠電泳分離。化學切割反應:包括鹼基的修飾修飾的鹼基從其糖環上轉移出去在失去鹼基的糖環處DNA斷裂。
Sanger法
就是利用一種DNA聚合酶來延伸結合在待定序列模板上的引物。直到摻入一種鏈終止核苷酸為止。每一次序列測定由一套四個單獨的反應構成,每個反應含有所有四種脫氧核苷酸三磷酸(dNTP),並混入限量的一種不同的雙脫氧核苷三磷酸(ddNTP)。由於ddNTP缺乏延伸所需要的3-OH基團,使延長的寡聚核苷酸選擇性地在G、A、T或C處終止。終止點由反應中相應的雙脫氧而定。每一種dNTPs和ddNTPs的相對濃度可以調整,使反應得到一組長几百至幾千鹼基的鏈終止產物。它們具有共同的起始點,但終止在不同的的核苷酸上,可通過高解析度變性凝膠電泳分離大小不同的片段,凝膠處理後可用X-光膠片放射自顯影或非同位素標記進行檢測。
測序方法
生成互相獨立的若干組帶放射性標記的寡核苷酸,每組寡核苷酸都有固定的起點,但卻隨機終止於特定的一種或者多種殘基上。
由於DNA上的每一個鹼基出現在可變終止端的機會均等,因此上述每一組產物都是一些寡核苷酸混合物,這些寡核苷酸的長度由某一種特定鹼基在原DNA全片段上的位置所決定。
在可以區分長度僅差一個核苷酸的不同DNA分子的條件下,對各組寡核苷酸進行電泳分析,只要把幾組寡核苷酸加樣於測序凝膠中若干個相鄰的泳道上,即可從凝膠的放射自影片上直接讀出DNA上的核苷酸順序。
自動測序法
基因分析儀(即DNA測序儀),採用毛細管電泳技術取代傳統的聚丙烯醯胺平板電泳,套用該公司專利的四色螢光染料標記的ddNTP(標記終止物法),因此通過單引物PCR測序反應,生成的PCR產物則是相差1個鹼基的3'末端為4種不同螢光染料的單鏈DNA混合物,使得四種螢光染料的測序PCR產物可在一根毛細管內電泳,從而避免了泳道間遷移率差異的影響,大大提高了測序的精確度。由於分子大小不同,在毛細管電泳中的遷移率也不同,當其通過毛細管讀數視窗段時,雷射檢測器視窗中的CCD(charge-coupleddevice)攝影機檢測器就可對螢光分子逐個進行檢測,激發的螢光經光柵分光,以區分代表不同鹼基信息的不同顏色的螢光,並在CCD攝影機上同步成像,分析軟體可自動將不同螢光轉變為DNA序列,從而達到DNA測序的目的。分析結果能以凝膠電泳圖譜、螢光吸收峰圖或鹼基排列順序等多種形式輸出。
它是一台能自動灌膠、自動進樣、自動數據收集分析等全自動電腦控制的測定DNA片段的鹼基順序或大小和定量的高檔精密儀器。PE公司還提供凝膠高分子聚合物,包括DNA測序膠(POP6)和GeneScan膠(POP4)。這些凝膠顆粒孔徑均一,避免了配膠條件不一致對測序精度的影響。它主要由毛細管電泳裝置、Macintosh電腦、彩色印表機和電泳等附屬檔案組成。電腦中則包括資料收集,分析和儀器運行等軟體。它使用最新的CCD攝影機檢測器,使DNA測序縮短至2.5h,PCR片段大小分析和定量分析為10~40min。
由於該儀器具有DNA測序,PCR片段大小分析和定量分析等功能,因此可進行DNA測序、雜合子分析、單鏈構象多態性分析(SSCP)、微衛星序列分析、長片段PCR、RT-PCR(定量PCR)等分析,臨床上可除進行常規DNA測序外,還可進行單核苷酸多態性(SNP)分析、基因突變檢測、HLA配型、法醫學上的親子和個體鑑定、微生物與病毒的分型與鑑定等。
一、準備工作
1.BigDye測序反應試劑盒主要試劑是BigDyeMix,內含PE專利四色螢光標記的ddNTP和普通dNTP,AmpliTaqDNApolymeraseFS,反應緩衝液等。
2.pGEM-3Zf(+)雙鏈DNA對照模板0.2g/L,試劑盒配套試劑。
3.M13(-21)引物TGTAAAACGACGGCCAGT,3.2μmol/L,即3.2pmol/μl,試劑盒配套試劑。
4.DNA測序模板可以是PCR產物、單鏈DNA和質粒DNA等。模板濃度應調整在PCR反應時取量1μl為宜。本實驗測定的質粒DNA,濃度為0.2g/L,即200ng/μl。
5.引物需根據所要測定的DNA片段設計正向或反向引物,配製成3.2μmol/L,即3.2pmol/μl。如重組質粒中含通用引物序列也可用通用引物,如M13(-21)引物,T7引物等。
6.滅菌去離子水或三蒸水。
7.0.2ml或和0.5ml的PCR管蓋體分離。
8.3mol/L醋酸鈉(pH5.2)稱取40.8gNaAc·3H2O溶於70ml蒸餾水中,冰醋酸調pH至5.2,定容至100ml,高壓滅菌後分裝。
9.70%乙醇和無水乙醇。
10.NaAc/乙醇混合液取37.5ml無水乙醇和2.5ml3mol/LNaAc混勻,室溫可保存1年。
11.POP6測序膠。
12.模板抑制試劑(TSR)。
13.10×電泳緩衝液。
14.全自動DNA測序儀。
15.PCR儀。
16.台式冷凍高速離心機。
17.台式高速離心機或袖珍離心機。
二、PCR測序反應
1.取0.2ml的PCR管,用記號筆編號,將管插在顆粒冰中,按下表加試劑:
所加試劑測定模板管標準對照管
BigDyeMix1μl1μl
待測的質粒DNA1μl-
pGEM-3Zf(+)雙鏈DNA-1μl
待測DNA的正向引物1μl-
M13(-21)引物-1μl
滅菌去離子水2μl2μl
總反應體積5μl,不加輕礦物油或石蠟油,蓋緊PCR管,用手指彈管混勻,稍離心。
2.將PCR管置於9600或2400型PCR儀上進行擴增。98℃變性2min後進行PCR循環,PCR循環參數為96℃10s,50℃5s,60℃4min,25個循環,擴增結束後設定4℃保溫。
三、醋酸鈉/乙醇法純化PCR產物
1.將混合物離心,將擴增產物轉移到1.5mlEP管中。
2.加入25μl醋酸鈉/乙醇混合液,充分振盪,置冰上10min以沉澱DNA。12000r/min於4℃離心30min,小心棄上清。
3.加70%(V/V)的乙醇50μl洗滌沉澱2次。12000r/min於4℃離心5min,小心棄上清和管壁的液珠,真空乾燥沉澱10~15min。
四、電泳前測序PCR產物的處理
1.加入12μl的TSR於離心管中,劇烈振盪,讓其充分溶解DNA沉澱,稍離心。
2.將溶液轉移至蓋體分離的0.2mlPCR管中,稍離心。
3.在PCR儀上進行熱變性(95℃2min),冰中驟冷,待上機。
五、上機操作
1.按儀器操作說明書安裝毛細管,進行毛細管位置的校正,人工手動灌膠和建立運行的測序順序檔案。
2.儀器將自動灌膠至毛細管,1.2kV預電泳5min,按編程次序自動進樣,再預電泳(1.2kV,20min),在7.5kV下電泳2h。
3.電泳結束後儀器會自動清洗,灌膠,進下一樣品,預電泳和電泳。
4.每一個樣品電泳總時間為2.5h。
5.電泳結束後儀器會自動分析或列印出彩色測序圖譜。
六、序列分析
儀器將自動進行序列分析,並可根據用戶要求進行序列比較。如測序序列已知,可通過序列比較以星號標出差異鹼基處,提高工作效率。
七、儀器清洗
測序完畢按儀器操作規程進行儀器清洗與保養。
八、計算
1.測序反應精確度計算公式:100%-差異鹼基數(不包括N數)/650×100%。
2.差異鹼基即測定的DNA序列與已知標準DNA序列比較不同的鹼基,N為儀器不能辨讀的鹼基。
非同位素銀染色法
SILVERSEQUENCETMDNA測序系統是一種無放射性的序列分析系統,它通過靈敏的銀染方法檢測凝膠中的條帶。銀染提供了一種對於放射性或螢光法來說更加快速,廉價的替代方法。測序結果可以在同一天內得到;電泳完成後經90分鐘就可讀序,這是常規的放射性測序法做不到的。此外,SILVERSEQUENCETM系統用未修飾的5'OH寡聚核苷酸作為引物,減少了特殊修飾寡聚核苷酸的花費。該系統不需要放射性方法中對同位素的謹慎操作,也不需要螢光法或化學發光技術的昂貴試劑。另外,也不需要象大多數螢光法那樣用儀器來檢測序列條帶。
TaqDNA聚合酶在95℃時極強的熱穩定性。本系統利用的測序級TaqDNA聚合酶是一種TaqDNA聚合酶的修飾產品,對於雙鏈DNA模板有非常好的效果,具有高度的準確性,能產生均一的條帶,且背景低。
SILVERSEQUENCETM系統包含被修飾的核苷酸混合物,如7-去氮dGTP(7-deazadGTP,或dITP)替代dGTP可清除由GC豐富區域所引起的條帶壓縮現象。
退火溫度是熱循環測序中最重要的因素。高退火溫度可減少模板二級結構。提高引物結合模板配對的嚴謹性。鏈重退火和模板二級結構則限制了小片斷PCR產物(<500bp)得到清楚的序列數據的能力。引物延伸起始於每個循環的退火階段。在較低溫度時,聚合酶可能會遇到堅固的二級結構區域,它可導致聚合酶解離。則在四個電泳道中均有同一相對位置的條帶。因為這些原因,應該使用儘可能高的退火溫度。對於有牢固二級結構的模板建議使用95℃變性、70℃退火/延伸的循環模式。一般來說,較長的引物及GC含量高的引物能得到較強的信號。實驗結果表明,>24mer的GC含量約為50%的引物可得到最佳結果。
由於本系統採用熱循環裝置,與常規的測序方法相比具有如下幾點好處:(1).本方法線性擴增模板DNA產生足夠的產物使銀染技術能夠檢測序列條帶,測序反應需要0.03~2pmol模板DNA,隨模板種類而定。(2).在每一個變性循環中的高溫可以取代對雙鏈DNA(dsDNA)模板的鹼變性及乙醇沉澱過程,變性循環也有助於消除由於線性dsDNA模板(如PCR反應產物)快速重退火所引起的問題。(3).高溫聚合酶反應減弱了DNA模板的二級結構,允許聚合酶穿過高度二級結構化的區域。
一、試劑準備
1.SILVERSEQUENCETMDNA測序試劑盒。
2.丙烯醯胺和甲叉雙丙烯醯胺儲備液(38%丙烯醯胺W/V,2%甲叉雙丙烯醯胺W/V):95g丙烯醯胺,5g甲叉雙丙烯醯胺溶於140ml雙蒸水中,定容至250ml,0.45mm過濾器過濾後,貯於棕色瓶中,置於4℃冰櫃可保存2周。
3.10%過硫酸銨,0.5g過硫酸銨溶於4ml水中,定容至5ml,應新配新用。
4.10×TBE緩衝液(1mol/LTris,0.83mol/L硼酸,10mmol/LEDTA):121.1gTris,51.35g硼酸,3.72gNa2EDTA·2H2O,溶於雙蒸水中定容至1升,置於4℃下可貯存2周,其pH約為8.3。
5.TBE電極緩衝液:10×TBE緩衝液稀釋至1×TBE備用。
6.TEMED
7.固定/停止溶液:10%冰醋酸(V/V)配製2升備用。
8.染色溶液:硝酸銀2g,甲醛3ml,溶於2升超純水中備用。
9.顯影溶液:60g碳酸鈉(Na2CO3)溶於2升超純水中,使用前加3ml37%甲醛和40ml硫代硫酸鈉溶液(10mg/ml)。
10.95%乙醇。
11.0.5%冰乙酸。
12.Sigmacote(SigmaCAT.#SL-2)。
二、測序反應
1.對於每組測序反應,標記四個0.5mleppendorf管(G、A、T、C)。每管加入2ml適當的d/ddNTP混合物(d/ddNTPMix)。各加入1滴(約20μl)礦物油,蓋上蓋子保存於冰上或4℃備用。
2.對於每組四個測序反應,在一個eppendorf管中混合以下試劑:
(1)樣品反應:
質粒模板DNA:2.1pmol
5×測序緩衝液:5ml
引物:4.5pmol
無菌ddH2O至終體積16ml
(2)對照反應
pGEM-3Zf(+)對照DNA(4mg):4.0ml
5×測序緩衝液:5ml
pUC/M13正向引物(4.5pmol):3.6ml
3.在引物/模板混合物(以上第2步)中加入1.0ml測序級TaqDNA聚合酶(5μ/ml)。用吸液器吸動幾次混勻。
4.從第3步的酶/引物/模板混合物中吸取4ml加入每一個d/ddNTP混合物的管內。
5.在微量離心機中離心一下,使所有的溶液位於eppendorf管底部。
6.把反應管放入預熱至95℃的熱循環儀,以【注意】中循環模式為基準,開始循環程式。對於每個引物/模板組合都必須選擇最佳退火溫度。下列程式一般能讀出從引物開始350鹼基的長度。
7.熱循環程式完成後,在每個小管內加入3μlDNA測序終止溶液,在微量離心機中略一鏇轉,終止反應。
【注意】
(1)測序所用模板DNA的量一般按下面要求加入:
模板種類/長度模板量
200bp(PCR產物):16ng(120fmol)
3000~5000bp(超螺鏇質粒DNA):4mg(2pmol)
48000bp(λ,粘粒DNA):1mg(31fmol)
由於超螺鏇質粒產生的信號比鬆弛的線性雙鏈DNA弱,因此使用超螺鏇質粒作為模板時其用量要比其它模板大一些。
(2)計算與4.5pmol相當的引物納克數可用以下一般公式:
4.5pmol=1.5ng×n,其中n為引物鹼基數
計算與1pmol相當的引物微克數可用以下一般公式:
dsDNA:1pmol=(6.6×10mg)×n,其中n為模板鹼基對數
ssDNA:1pmol=(3.3×10mg)×n,其中n為模板鹼基數
(3)為阻止TaqDNA聚合酶延伸非特異性退火引物,熱循環儀必須預熱至95℃。溫度變換應越快越好。下面的循環時間不包括變溫時間。如果你無法確定使用何種模式,建議從模式1開始。
模式1:適用於引物<24鹼基或GC含量<50%
95℃2分鐘,然後:95℃30秒(變性),42℃30秒(退火),70℃1分鐘(延伸)。
模式2:適用於≥24鹼基或略短的GC含量≥50%的引物。
95℃2分鐘,然後:95℃30秒(變性),70℃30秒(退火/延伸)。
(4)在加入終止溶液之後樣品可在4℃保存過夜。
三、測序凝膠板的製備
1.玻璃板的處理
銀染測序的玻璃板一定要非常清潔,一般先用溫水和去污劑洗滌,再用去離子水沖洗玻璃板,除去殘留的去污劑,最後用乙醇清洗玻璃板。玻璃板上遺留的去污劑微膜可能導致凝膠染色時背景偏高(棕色)。短玻璃板經粘合溶液處理可將凝膠化學交聯於玻璃板上。這一步對於在銀染操作過程中防止凝膠撕裂至關重要。
(1)短玻璃板的處理
①在1ml95%乙醇,0.5%冰乙酸中加入5ml粘合矽烷(BindSilane),配成新鮮的粘合溶液。
②用經浸透新配的粘合溶液浸透的吸水棉紙擦拭仔細清洗過並已經自然乾燥的玻璃板,整個板面都必須擦拭。
③4~5分鐘後,用95%乙醇單向擦玻璃板,然後略用力沿垂直方向擦拭。重複三次這一清洗過程,每次均須換用乾淨的紙,除去多餘的粘合溶液。
【注意】
①在95%乙醇單向擦玻璃板時過度用力會帶走過多的粘合矽烷,使凝膠不能很好地粘附。
②準備長玻璃板之前要更換手套,防止粘染粘合矽烷。
③防止粘合溶液沾染在長玻璃板上是很重要的,否則將導致凝膠撕裂。
(2)長玻璃板的處理:
①用浸透Sigmacote溶液的棉紙擦拭清洗過的長玻璃板。
②5~10分鐘後用吸水棉紙擦拭玻璃板以除去多餘的Sigmacote溶液。
【注意】
①用過的凝膠可在水中浸泡後用剃鬚刀片或塑膠刮刀颳去。玻璃板須用去污劑完全清洗。或者凝膠用10%NaOH浸泡後除去。為防止交叉污染,用於清洗短玻璃板的工具必須與清洗長玻璃板的工具分開,如果出現交叉污染,以後製備的凝膠可能撕裂或變得鬆弛。
2.凝膠的製備
(1)玻璃板經粘合矽膠和Sigmacote處理後,即可固定玻璃板。該方法是用0.2mm或0.4mm厚的邊條置於玻璃板左右兩側,將另一塊玻璃板壓於其上。在長玻璃板的一側插入鯊魚齒梳平的一面邊緣,用夾子固定住。
(2)根據所需要的凝膠濃度,按下表製備測序凝膠,一般6%~8%的膠濃度可獲得較好的結果。配製過程中,先用適量雙蒸水溶解尿素,再加入Acr&Bis和10×TBE緩衝液,再用雙蒸水調終體積至99.2ml,並用0.45mm的濾膜過濾,然後加過硫酸銨和TEMED。溶解尿素時不必加熱。如果確需加熱則應等溶液完全冷卻後,方可加入TEMED和過硫酸銨。一般在膠灌制後4~6分鐘,即開始聚合,如果聚合不好,則應使用高濃度的TEMED和過硫酸銨。
(3)膠配製好後,即可灌制膠板。一般是將凝膠沿著壓條邊緣緩慢地倒入玻璃板的槽中,倒完後,靜止放置使之聚合完全。
【注意】
①使用夾子固定玻璃板時,最好夾子的力量稍大一些,防止因力量不足使灌膠的過程中出現漏膠液現象。
②灌制凝膠的過程中要嚴防產生氣泡,否則影響測序的結果。
四、電泳
1.預電泳
(1)當凝膠聚合完全後,撥出鯊魚齒梳,將該梳子反過來,把有齒的一頭插入凝膠中,形成加樣孔。
(2)立即將膠板固定在測序凝膠槽中,一般測序凝膠槽的上下槽是分開的,因而只有在固定好凝膠板後,方能加入TBE緩衝液。
(3)稀釋10×TBE緩衝液至1×TBE,將該緩衝液加入上下二個電泳槽中,去除產生的氣泡,接上電源準備預電泳。
(4)有些電泳槽,如LKB的Macrophor等是使用水浴加熱的,則應先將水浴加熱至55℃後進行預電泳。有的不使用水浴加熱,依靠電泳過程中自身產生的熱進行保溫,如上海求精有機玻璃儀器生產的測序電泳槽,這種槽需夾上二塊散熱鋁板,使整個凝膠板的溫度一致。
(5)按30V/cm的電壓預電泳20~30分鐘。預電泳的過程是去除凝膠的雜質離子,同時使凝膠板達到所需的溫度。高溫電泳可防止GC豐富區形成的髮夾狀結構,影響測序的結果。
【注意】
①用鯊魚齒梳製作加樣孔時,應注意將齒尖插入膠中0.5mm左右,千萬注意不能使加樣孔滲漏,否則得不到正確的結果。
②應時刻注意上面電泳槽中的緩衝液是否滲漏,否則極易造成短路而損壞電泳儀。
2.樣品的製備
當在預電泳時,即可進行樣品的製備,將反應完畢的樣品在沸水浴中加熱1~3分鐘,立即置於冰上即可。如果樣品長時間不用,則應重新處理。可使用4~6%聚丙烯醯胺凝膠,膠厚0.4mm。厚度小於0.4mm的膠可能導致信號太弱。加樣時不必吸去上層覆蓋的礦物油,但要小心地吸取礦物油下的藍色樣品。
3.上樣及電泳
關閉電泳儀,用移液槍吸緩衝液清洗樣品孔,去除在預電泳時擴散出來的尿素,然後立即用毛細管進樣器吸取樣品,加入樣品孔中。上樣順序一般為G、A、T、C。加樣完畢後,立即電泳。開始可用30V/cm進行電泳,5分鐘後可提高至40~60V/cm,並保持恆壓狀態。一般來說,一個55cm長,0.2mm厚的凝膠板,在2500V恆壓狀態下電泳2小時即可走到底部,同時在電泳過程中,電流可穩定地從28mA降至25mA。為了能讀到更長的序列,可採用兩輪或多輪上樣。
【注意】
①上樣電泳時,一定要注意凝膠板的溫度是否達到55℃左右,如果還沒有達到,則應等溫度達到後才能上樣電泳。
②一般來說電泳時,不宜使用太高的電壓,因為太高的電壓會使凝膠的解析度降低,並且使帶擴散。電泳中可進行恆功率電泳。
五、測序凝膠的銀染
染色過程要求凝膠浸在塑膠盤中。因而至少使用兩個盤子,大小與玻璃板類似。在盤中加入新鮮溶液之前須用高質量的水洗滌盤子。
1.電泳完畢後用一個塑膠片子小心地分開兩板,凝膠應該牢固地附著在短玻璃板上。
2.固定凝膠:將凝膠(連玻璃板)放入塑膠盤,用固定/停止溶液浸沒,充分振盪20分鐘或直至樣品中染料完全消失,膠可在固定/停止溶液中保存過夜(不振盪)。保留固定/停止溶液,用於終止顯影反應。
3.洗膠:用超純水振盪洗膠3次,每次2分鐘。從水中取出,當轉移至下一溶液時拿著膠板邊沿靜止10~20秒,使水流盡。
4.凝膠染色:把凝膠移至染色溶液充分搖動30分鐘。
5.凝膠顯影
(1)在顯影溶液中加入甲醛(3ml)和硫代硫酸鈉溶液(400μl)以完成顯影液的配製。
(2)從染色溶液中取出凝膠放入裝有超純水的盤中浸洗5~10秒。注意,把凝膠從超純水轉移到顯影溶液的總時間不能長於5~10秒。浸泡時間過長則導致信號微弱或喪失信號。若浸泡時間過長,可重複第五步用染色液浸泡。
(3)立刻將凝膠轉移至1升(總量的一半)預冷的顯影液充分振盪直至模板帶開始顯現或開始出現第一批條帶,把凝膠移入剩下的1升顯影液中繼續顯影2~3分鐘或直至所有條帶出現。
6.固定凝膠:在顯影液中直接加入等體積的固定/停止溶液。停止顯影反應,固定凝膠。
7.在超純水中浸洗凝膠兩次,每次2分鐘,注意在本操作中戴手套拿著膠板邊緣避免在膠上印上指紋。
8.將凝膠置於室溫乾燥或用抽氣加熱法乾燥。在可見光燈箱或亮白,黃色背景(如紙)上觀察凝膠,若需永久保存的記錄,則可用EDF膠片保留實驗結果。
【注意】
測序產物的銀染是顯現序列信息的一種新方法,本系統的成敗受幾個因素的影響
①水的質量對於染色的成功極其重要。超純水(NANOpureR或Milli-QR的水)或雙蒸水可獲得較好的效果,如果水中有雜質,則低分子量條帶可能無法出現。
②碳酸鈉也非常重要。使用新鮮的,美國化學學會級碳酸鈉較好,如Fisher和KodakACS試劑級碳酸鈉(FisherCat#S263-500或S262-3,或KodakCat#109-1990),一般可獲得較好的結果。
③色後的洗滌步驟是非常關鍵的。如果凝膠洗滌時間太長,銀顆粒會脫離DNA,產生很少或沒有序列信號。如果洗滌時間過長,染色步驟可以重新進行。
④如果凝膠厚度超過0.4mm或丙烯醯胺濃度高於4~6%,則有必要延長固定和染色的時間。如果凝膠比0.4mm薄,染色反應後的洗滌必須縮短至不超過5秒。
⑤在室溫下進行所有步驟,顯影反應除外。顯影溶液必須預冷至10~12℃以減小背景雜色。注意:臨用前在顯影溶液中加入甲醛和硫代硫酸鈉。用新配的染色及顯影溶液。不要重複使用任何溶液。
六、EDF膠片顯影
使用EDF膠片可增強測序條帶的對比度,如果測序膠上條帶很淡,我們建議把數據轉移至EDF膠片,銀染膠在其影像轉移至EDF膠片之後可增強條帶可讀性。
1.在暗室內,將染色過的粘於玻璃板的凝膠(膠面向上)置於螢光燈箱上。如有合適的漫射板,亦可用白燈箱,為確保曝光時間,用一小條EDF膠片曝光不同時間,檢查不同的曝光強度,一般曝光20~40秒可得較好結果。
2.在紅燈下找到EDF膠片有缺刻的一角,然後把膠片置於凝膠上,使缺口位於左上角。由於EDF膠片是單面的,因此必須確保缺口在左上角。
3.在EDF膠片上放置一乾淨、乾燥的玻璃板,開亮燈箱約20秒。
4.用沖顯放射自顯影膠片的步驟手工顯影EDF膠片,可使用下列操作過程:
(1)在KodakGBX顯影液中顯影1~5分鐘;
(2)水洗1分鐘;
(3)在KodakGBX定影液中定影3分鐘;
(4)水洗1分鐘。
【注意】
①進行EDF膠片顯影之前凝膠必須完全乾燥。戴手套進行操作,以免印上指紋。同時注意EDF膠片不能用自動膠片處理器。
②對於不同的光源最佳曝光時間可能不同,通過對一小條EDF膠片曝光不同時間以選擇你所用光源的最佳曝光時間,參閱膠片說明書。
③曝光時間短則EDF膠片影像較深,曝光時間長則有助於減弱背景。
鳥槍法
“鳥槍法”是一種由生物基因組提取目的基因的方法。首先利用物理方法(如剪下力、超音波等)或酶化學方法(如限制性內切核酸酶)將生物細胞染色體DNA切割成為基因水平的許多片段,繼而將這些片段與適當的載體結合,將重組DNA轉入受體菌擴增,獲得無性繁殖的基因文庫,再結合篩選方法,從眾多的轉化子菌株中選出含有某一基因的菌株,從中將重組的DNA分離、回收。
這種方法也就是套用基因工程技術分離目的基因,其特點是繞過直接分離基因的難關,在基因組DNA文庫中篩選出目的基因。可以說這是利用“溜散彈射擊”原理去“命中”某個基因。由於目的基因在整個基因組中太少太小,在相當程度上還得靠“碰運氣”,所以人們稱這個方法為“鳥槍法”或“散彈槍”實驗法。
一、用限制酶切下目的片段的大片段InsertDNA,並用Agarose凝膠電泳進行回收。
二、對回收的大片段DNA用物理方法(如超音波等)進行切斷處理,然後用T4DNAPolymerase對小片段DNA進行末端平滑化。
三、進行Agarose電泳,切膠回收1kbp~2kbp的小片段DNA。然後用BcaBestDNAPolymerase在DNA的3'端加上一個A鹼基。
四、把加上A-tail的DNA片段連入T-Vector中,然後轉化,挑選克隆。
五、對陽性克隆(含1kbp~2kbpInsert)進行DNA測序。
六、對數據進行編輯,最後連成一條大片段的DNA序列。
測序技術
高通量測序技術(High-throughputsequencing)又稱“下一代”測序技術("Next-generation"sequencingtechnology),以能一次並行對幾十萬到幾百萬條DNA分子進行序列測定和一般讀長較短等為標誌。
根據發展歷史、影響力、測序原理和技術不同等,主要有以下幾種:大規模並行簽名測序(MassivelyParallelSignatureSequencing,MPSS)、聚合酶克隆(PolonySequencing)、454焦磷酸測序(454pyrosequencing)、Illumina(Solexa)sequencing、ABISOLiDsequencing、離子半導體測序(Ionsemiconductorsequencing)、DNA納米球測序(DNAnanoballsequencing)等。
MPSS
由LynxTherapeutics公司在90年代發展的MassivelyParallelSignatureSequencing,MPSS技術是“下一代”測序技術發展的先驅。MPSS是一種基於磁珠(bead)和接頭(adaptor)連線和解碼的複雜技術,測定結果短,多用於轉錄組測序,測定基因表達量。MPSS測定結果有序列偏好性而易丟失DNA中某些特定序列,且操作複雜,已逐漸淡出,被新的方法替代。
PolonySequencing
2005年哈佛GeorgeChurch實驗室發展起來的,基於乳化PCR(emulsionPCR)和自動顯微鏡等技術的測序方法。相關技術已整合進ABI公司的SOLiD測序技術平台。
454焦磷酸測序
由454公司發展的並行焦磷酸測序方法。該方法在油溶液包裹的水滴中擴增DNA(即emulsionPCR),每一個水滴中開始時僅包含一個包被大量引物的磁珠和一個連結到微珠上的DNA模板分子(控制DNA濃度出現的大機率事件)。將emlusionPCR產物載入到特製的PTP板上,板上有上百萬個孔,每個微孔只能容納一個磁珠。DNAPolymerase在將一個dNTP聚合到模板上的時候,釋放出一個PPi(焦磷酸分子);在ATP-Sulfurylase(ATP硫酸化酶)催化下,PPi與APS生成一個ATP分子;ATP分子在Luciferase(螢光素酶)的作用下,將luciferin(螢光素)氧化成oxyluciferin,同時產生的可見光被CCD光學系統捕獲,獲得一個特異的檢測信號,信號強度與相應的鹼基數目成正比。通過按順序分別並循環添加四種dNTP,讀取信號強度和發生時間,實現DNA序列測定。這一技術的讀長和每一鹼基耗費都介於Sanger法測序和Solexa和SOLiD方法之間。454公司現下屬於Roche公司。
Illumina(Solexa)sequencing
Solexa公司開發了一種可反轉的染料終止法。DNA模板首先連線到與固體介質(如玻璃板)交聯的引物上,並擴增形成本地微克隆。四種ddNTPs依次添加到體系中,未結合的ddNTPs在添加下一種核苷酸前被沖洗走。與454的焦磷酸測序不同,這種方法一次只延伸一個鹼基。
用途
在基礎生物學研究中,和在眾多的套用領域,如診斷,生物技術,法醫生物學,生物系統學中,DNA序列知識已成為不可缺少的知識。具有現代的DNA測序技術的快速測序速度已經有助於達到測序完整的DNA序列,或多種類型的基因組測序和生命物種,包括人類基因組和其他許多動物,植物和微生物物種的完整DNA序列。
個人DNA測序
基因測序最廣為人知的,是影星安吉麗娜·朱莉通過基因檢測,選擇手術切除乳腺以降低患乳腺癌風險。2011年去世的蘋果公司創始人史蒂夫·賈伯斯患癌時,也曾接受過全基因測序。
在中國,多家醫療機構、體檢中心,甚至公司提供的體檢套餐,內容覆蓋腫瘤、高血壓、糖尿病等基因檢測,價格從數百元到數萬元不等。
2014年2月,國家食藥監管總局、國家衛計委聯合發出通知,要求在相關準入標準、管理規範出台以前,任何醫療機構不得開展基因測序臨床套用;已經開展的,要立即停止。
2016年8月,NASA太空人凱特•魯賓斯在國際空間站內成功完成微重力條件下的DNA測序,這標誌著人類已迎來“能對太空活體生物進行基因測序”的全新時代。
