
成份
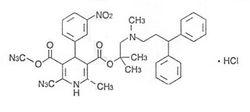
化學名稱:3,5-吡啶二羧酸,1,4-二氫-2,6-二甲基-4-(3-硝基苯)2-[(3,3-二苯基丙基)甲胺]-1,1-二甲基甲酯鹽酸鹽。
化學結構式:
 鹽酸樂卡地平片
鹽酸樂卡地平片分子式:CHNO·HCl
分子量:648.2
性狀
本品為淡黃色,中間有刻痕的圓形薄膜衣片。
適應症
本品治療輕、中度原發性高血壓。
規格
10mg/片
用法用量
用法
每天1次,餐前15分鐘口服。
用量
推薦劑量為10 mg,根據病人的個體反應可增至每次20 mg。
不良反應
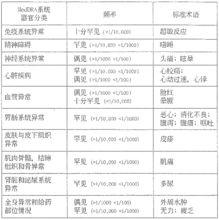
大約1.8%接受治療的患者會發生不良反應。下表顯示的是根據MedDRA系統器官分類體系歸類的按發生頻率分級(偶見,罕見)的至少可能存在因果關係的藥物不良反應的發生率。
根據表中所示,臨床對照試驗中報導的最常發生且發生在不足1%患者中的藥物不良反應是頭痛、眩暈、外周水腫,心動過速,心悸和臉紅。
 鹽酸樂卡地平片
鹽酸樂卡地平片在上市後套用中,下列不良反應的自發報告非常罕見(<l/10,000):齒齦增生,可逆的血清肝轉氨酶升高,低血壓,尿頻和胸痛。
在一些罕見的情況下,部分二氫吡啶類藥物可導致心前區疼痛或者心絞痛。極少數先前存在心絞痛的患者,其心絞痛發作頻率、持續時間或者發作的嚴重程度會增加。還有心肌梗死的個例報導。
樂卡地平不會對血糖和血脂水平產生不利影響。
禁忌
·對活性成分“樂卡地平”、任何二氫吡啶類或者任何藥物賦形劑過敏
·妊娠與哺乳期(見【孕婦及哺乳期婦女用藥】)
·有生育可能的婦女,除非採取了有效的避孕方
·左室流出道梗阻
·未治療的充血性心衰
·不穩定型心絞痛
·重度肝腎功能損害
·心肌梗死一個月內
·同時服用CYP3A4的強效抑制劑、環孢素或柚子汁(見【藥物相互作用】)
注意事項
病態竇房結綜合徵的患者(如果體內沒有安裝起搏器),在套用樂卡地平時應被密切觀察。儘管血流動力學的對照研究並沒有發現本品對心室功能的損害,但左室功能不全的患者套用本品時仍需小心。有證據提示缺血性心臟病患者套用短效二氫毗啶類藥物時可能會增加心血管風險。儘管樂卡地平是長效藥物,但在這些患者中套用也應注意。
在一些罕見的情況下,部分二氫吡啶類藥物會導致心前區疼痛或者心絞痛,極少數先前存在心絞痛的患者,其心絞痛發作頻率、持續時間或嚴重程度會增加。還有心肌梗死的個例報導(見【不良反應】)。
肝腎功能異常時的套用:輕到中度肝或腎功能異常患者在開始本品治療時應謹慎。儘管這類人群可能耐受常用的推薦劑量,但將日劑量增至20mg時則需要注意。由於肝功能受損時抗高血壓效果可能會增強,因此需要考慮調整劑量。
不推薦在肝功能重度損害或者重度腎功能不全 (GFR<30ml/min)的患者中套用樂卡地平。
服藥期間應避免飲酒或含酒精的飲料,因為這可能會增加抗高血壓藥物的血管擴張作用(見【藥物相互作用】)。
CYP3A4酶的誘導劑,如抗癲癇藥物(苯妥英,卡馬西平等)和利福平,可能降低樂卡地平的血漿水平,因此樂卡地平的有效性可能被降低(見【藥物相互作用】)。
每片藥物含30mg乳糖,因此不能套用於Lapp乳糖酶不足,半乳糖血症或者葡萄糖/半乳糖吸收不良綜合徵患者。
孕婦及哺乳期婦女用藥
大鼠和兔動物實驗研究的數據表明,樂卡地平無致畸作用,而且大鼠的生育能力也並未受到損害。但是,由於尚無樂卡地平套用於妊娠與哺乳期的臨床經驗,而且其他二氫吡啶類藥物在動物中發現了致畸作用,因此樂卡地平不能套用於妊娠期婦女,育齡期婦女需要採取有效的避孕方式後方能使用本品。由於樂卡地平具有高親脂性,乳汁中可能會有分布。因此,哺乳期婦女不能服用本品。
兒童用藥
18歲以下患者不得服用。
老年用藥
對老年患者一般無需做特別的劑量調整,但在治療開始時應予以關注。
藥物相互作用
已知樂卡地平經CYP3A4酶代謝,因此同時服用CYP3A4酶的抑制劑和誘導劑會影響樂卡地平的代謝和清除。應避免同時處方樂卡地平和CYP3A4酶抑制劑(如:酮康唑、伊曲康唑、利托那韋、紅黴素、竹桃黴素) (見【禁忌】)。
一項與CYP3A4酶強阻滯劑一酮康唑的相互作用研究表明,血漿樂卡地平的濃度水平顯著增加(優性異構體S—樂卡地平的曲線下面積(AUC)增加了15倍,峰值藥物濃度Cmax增加了8倍)。
環孢素和樂卡地平不能一起套用(見【禁忌】)。
已發現兩者一起套用後,樂卡地平和環孢素的血漿濃度均增加。一項在年輕健康志願者中的研究表明,當服用樂卡地平3小時後再服用環孢素時,樂卡地平的血漿濃度並無改變,而環孢素的AUC增加了27%。但是如果同時服用樂卡地平和環孢素,則樂卡地平的血藥濃度升高了3倍,環孢素的AUC增加了21%。樂卡地平不能與柚子汁同服(見【禁忌】)。
同其他二氫吡啶類藥物一樣,樂卡地平對於柚子汁的代謝抑制作用敏感,會導致全身性利用度的提高從而增加降壓效果。
老年志願者同時口服20mg樂卡地平與咪達唑侖時,樂卡地平的吸收會增加(約40%),而吸收速率會下降(tmax從1.75小時延長到3小時)。咪達唑侖濃度無變化。
同時處方樂卡地平與其他CYP3A4酶底物,如特非那定、阿司咪唑、III類抗心律失常藥物如胺碘酮、奎尼丁等時應當謹慎。
同時套用樂卡地平和CYPSA4酶的誘導劑,如抗癲癇藥物(苯妥英,卡馬西平等)和利福平時應當注意,因為降壓效果可能會降低,應當比平時更頻繁地監測患者的血壓。
樂卡地平和美托洛爾(一種主要通過肝臟清除的"—腎上腺素受體阻滯劑)同時套用時,美托洛爾的生物利用度無明顯變化,但樂卡地平的生物利用度下降了50%。這種效應可能是由於β—腎上腺素受體阻滯劑減少了肝臟的血流,因而也可能會對這一類其它藥物有類似的作用。因此,樂卡地平可以安全地與β—腎上腺素受體阻滯劑同時服用,但是可能需要調整劑量。
一項在6s±7歲(平均年齡±標準差)的志願者中進行的與氟西汀(一種CYP2D6和CYP3A4酶的抑制劑)的相互作用研究表明,樂卡地平的藥代動力學未發生臨床相關變化。
同時服用日劑量為800mg的西咪替丁不會引起樂卡地平血藥濃度的顯著改變,但是如果服用更大劑量時則需謹慎,因為樂卡地平的生物利用度和降壓作用可能會增加。
長期服用β—甲基地高辛的患者同時服用20mg樂卡地平,兩種藥物之間未見藥代動力學相互作用。健康志願者在服用地高辛後再空腹服用20mg樂卡地平顯示:地高辛的Cmax增加了33%,但是AUC和腎臟清除率無顯著變化。同時服用地高辛的患者應密切監測地高辛中毒的臨床徵象。
20mg樂卡地平和40mg辛伐他汀多次合用時,樂卡地平的AUC無明顯改變,但是辛伐他汀的AUC增加56%,並且它的活性產物β—羥酸增加了28%,只是這些改變不太可能具有臨床相關性。如果按照藥物的使用方法,樂卡地平在上午口服而辛伐他汀在晚上口服,這種相互作用就不會發生。
健康志願者空腹時同時服用20mg樂卡地平和華法林並不改變華法林的藥代動力學。
樂卡地平可以安全地與利尿藥和血管緊張素轉化酶抑制劑同時套用。
服藥期間應避免飲酒或含酒精的飲料,因為這可能會增加抗高血壓藥物的血管擴張作用(見【注意事項】)。
藥物過量
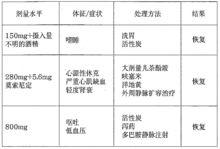
在上市後套用中曾有三例藥物過量的病例報導(每例分別服用了150mg、280mg和800mg樂卡地平以試圖自殺)。
 鹽酸樂卡地平片
鹽酸樂卡地平片同其他二氫吡啶類一樣,藥物過量預計會產生周圍血管過度擴張而出現顯著的低血壓和反射性心動過速。在嚴重低血壓,心動過緩和意識喪失的情況下,心血管支持將有助於治療,心動過緩時可靜脈套用阿托品。
考慮到樂卡地平較長的藥理學效應,需要對服藥過量的患者監測心血管情況至少24小時。目前尚無評價透析效果的資料。由於該藥物具有高親脂性,因此藥物血漿水平對藥物過量危險期持續時間無指導作用,而且透析可能無效。
藥理毒理
藥理作用
樂卡地平是新一代的二氫吡啶類鈣通道組滯劑,具有較強的血管選擇性,起效平緩,降壓作用強,作用時間長,負性肌力作用少等特點。體外研究發現,樂卡地平對血管平滑肌有直接的舒張作用,因而在體內具有較強的降壓作用,但對心率和心輸出量的影響較小。由於具有較大的疏水基因,脂溶性強,樂卡地平進入體內後迅速分布至組織器官中,與血管平滑肌細胞膜結合緊密,釋放緩慢,所以,雖然該藥血清消除半衰期短,但作用持久。
毒性研究
動物的毒理學研究表明,治療劑量對自主神經系統、中樞神經系統或胃腸道功能沒有影響。雄性和雌性小鼠口服本品的LD分別為622 mg/kg和438 mg/kg。慢性毒性研究表明,大鼠口服1.5-120 mg/kg/天或狗口服1.5-30 mg/kg/天共52周,無明顯毒性作用產生,此結果與其他二氫吡啶類鈣拮抗劑相似。動物試驗未發現本品有致畸、致癌和致突變作用。
藥代動力學
口服10-20mg樂卡地平後,藥物被完全吸收。血藥峰值水平分別為3.30ng/ml±2.09s.d.和7.66ng/m1± 5.90s.d.,達峰時間一般為服藥後1.5~3小時。
樂卡地平的兩種異構體的血藥水平相似:血漿達峰時間相同,峰值水平和曲線下面積則S—異構體平均高出1.2倍。兩種異構體的清除半衰期基本一致,未觀察到兩種異構體在體內的相互轉化。
由於較高的首過代謝,患者進食後服用樂卡地平的絕對生物利用度約為l0%,然而健康志願者在空腹狀態下給藥的絕對生物利用度下降到前者的l/3。
高脂餐後2小時內口服樂卡地平,其生物利用度將增加4倍。因此,應在餐前服用樂卡地平。
樂卡地平從血漿到組織和器官的分布是迅速而廣泛的。
樂卡地平和血清蛋白的結合率超過了98%。由於重度肝腎功能異常的患者血漿蛋白水平會下降,因此樂卡地平在這些患者中的游離型藥物水平就會增加。
樂卡地平完全通過CYP3A4酶代謝;在尿液和糞便中沒有發現藥物的原型成分。藥物主要轉化為無活性的代謝產物,約50%從尿液中排泄。
對人體肝臟微粒體的體外研究表明,當濃度分別高於口服20mg樂卡地平的血漿峰濃度的160倍和40倍時,對CYP3A4和CYP2D6均有一定程度的抑制作用。
另外,人體的藥物相互作用研究顯示,樂卡地平並不改變咪達唑侖和美托洛爾的血漿水平,前者是一種典型的CYP3A4底物,而後者是一種典型的CYP2D6底物。因此治療劑量下的樂卡地平不會抑制被CYP3A4和CYP2D6代謝的藥物的生物轉化。
樂卡地平主要通過生物轉化清除。
樂卡地平的終末消除半衰期為8~10小時,由於其對脂膜的高結合性,治療作用會持續24小時。重複給藥未發現蓄積。
口服給藥時的劑量與樂卡地平血漿濃度不成正比(非線性動力學)。口服l0mg、20mg和40mg樂卡地平,觀察到的血漿峰濃度比為l∶3∶8,血藥濃度一時間曲線下面積比為l∶4∶18。表明首過代謝是逐漸飽和的。因此,利用度隨劑量的增加而增加。
對於老年患者和輕中度肝腎功能損害的患者,樂卡地平的藥代動力學表現與在普通人群中觀察到的相似。重度腎功能不全或者依賴於透析的患者顯示出較高的藥物水平(約70%)。
對於中重度肝功能損害的患者,樂卡地平的全身性生物利用度可能會增加,因為藥物主要是通過肝臟代謝。
貯藏
應將本品置於室溫(10-30℃)乾燥處及兒童接觸不到的地方。
包裝
鋁/不透明的PVC泡罩包裝。7,14,28,50片/盒。
有效期
36個月
執行標準
進口藥品註冊標準:JX20000320
