
定義
化合物導電的前提:其內部存在著自由移動的陰陽離子。
離子化合物在水溶液中或熔化狀態下能導電;共價化合物:某些也能在水溶液中導電。
導電的性質與溶解度無關,強電解質一般有:強酸強鹼,大多數鹽;弱電解質一般有:(水中只能部分電離的化合物)弱酸,弱鹼。另外,水是極弱電解質。
電解質不一定能導電,而只有在溶於水或熔融狀態是電離出自由移動的離子後才能導電
能導電的不一定是電解質判斷某化合物是否是電解質,不能只憑它在水溶液中導電與否,還需要進一步考察其晶體結構和化學鍵的性質等因素。例如,判斷硫酸鋇、碳酸鈣和氫氧化鐵是否為電解質。硫酸鋇難溶於水(20℃時在水中的溶解度為2.4×10-4g),溶液中離子
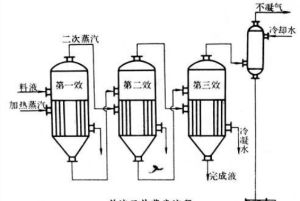 電解質溶液的蒸發
電解質溶液的蒸發 濃度很小,其水溶液不導電,似乎為非電解質。但溶於水的那小部分硫酸鋇卻幾乎完全電離(20℃時硫酸鋇飽和溶液的電離度為97.5%)。因此,硫酸鋇是電解質。碳酸鈣和硫酸鋇具有相類似的情況,也是電解質。從結構看,對其他難溶鹽,只要是離子型化合物或強極性共價型化合物,儘管難溶,也是電解質。
氫氧化鐵的情況則比較複雜,Fe3+與OH-之間的化學鍵帶有共價性質,它的溶解度比硫酸鋇還要小(20℃時在水中的溶解度為9.8×10-5g);而落於水的部分,其中少部分又有可能形成膠體,其餘亦能電離成離子。但氫氧化鐵也是電解質。
判斷氧化物是否為電解質,也要作具體分析。非金屬氧化物,如SO2、SO3、P2O5、CO2等,它們是共價型化合物,液態時不導電,所以不是電解質。有些氧化物在水溶液中即便能導電,但也不是電解質。因為這些氧化物與水反應生成了新的能導電的物質,溶液中導電的不是原氧化物,如SO2本身不能電離,而它和水反應,生成亞硫酸,亞硫酸為電解質。金屬氧化物,如Na2O,MgO,CaO,Al2O3等是離子化合物,它們在熔化狀態下能夠導電,因此是電解質。
可見,電解質包括離子型或強極性共價型化合物;非電解質包括弱極性或非極性共價型化合物。電解質水溶液能夠導電,是因電解質可以離解成離子。至於物質在水中能否電離,是由其結構決定的。因此,由物質結構識別電解質與非電解質是問題的本質。
另外,有些能導電的物質,如銅、鋁等不是電解質。因它們並不是能導電的化合物,而是單質,不符合電解質的定義。
電解質溶液的導電機理與金屬的導電機理不同。金屬是依靠自由電子的定向運動而導電,因而稱為電子導體,除金屬外,石墨和某些金屬氧化物也屬於電子導體。這類導體的特點是當電流通過時,導體本身不發生任何化學變化。電解質溶液的導電則依靠離子的定向運動,故稱為離子導體。但這類導體在導電的同時必然伴隨著電極與溶液界面上發生的得失電子反應:一般而言,陰離子在陽極上失去電子發生氧化反應,失去的電子經外線路流向電源正極;陽離子在陰極上得到外電源負極提供的電子發生還原反應。只有這樣整個電路才有電流通過。並且迴路中的任一截面,無論是金屬導線、電解質溶液,還是電極與溶液之間的界面,在相同時間內,必然有相同的電量通過。
基本特性
電解質溶液是指溶質溶解於溶劑後完全或部分解離為離子的溶液.相應溶質即為電解質.某物質是否為電解質並不是絕對的.同一物質在不同的溶劑中,可以表現出完全不同的性質.例如HCl在水中是電解質,但在苯中則為非電解質;葡萄糖在水中是非電解質,而在液態HF中卻是電解質.因此在談到電解質時決不能離開溶劑.一般把完全解離的電解質稱為強電解質,部分解離的電解質稱為弱電解質.這種分類方法只是為了討論問題的方便,並沒有反映出電解質的本質.原因是電解質的強弱隨環境而變.例如乙酸在水中為弱電解質,而在液氨中則為強電解質.LiCl和KI都是離子晶體,在水中為強電解質,而在醋酸或丙酮中都變成了弱電解質.在電化學中套用最廣泛的電解質溶液是電解質水溶液,本節主要討論電解質水溶液的基本特性.
正負離子
電解質溶液中的離子之間,除了具有像中性分子之間的那種相互作用之外,根據庫侖定律,還存在著靜電相互作用,即同性離子相互排斥,異性離子相互吸引.由分子運動論,兩個中性分子之間的相互吸引力近似地與兩粒子間距離的7次方成反比,而兩個異性離子之間的靜電吸引力卻與兩離子間距離的2次方成反比.這說明中性分子間的力為短程力,而帶電離子間的靜電引力為長程力.當電解質溶液較稀時,離子之間的距離較遠,各種近程力的作用可以略去不計,而長程力卻不可忽略.正是由於異性離子之間長程靜電引力的存在,使得電解質溶液即使在很稀時仍表現出對理想溶液的熱力學性質有較大的偏差.離子的靜電相互作用的強弱除與離子間的距離(溶液的濃度)有關外,還與溶劑的介電常數,離子的結構,大小,電荷,溶劑化程度等因素有關.
正負離子之間的庫侖引力,有可能使它們產生締合作用.當電荷相反的離子接近到一定距離時,若它們之間的靜電吸引勢能會遠遠大於熱運動動能,則在溶液中形成離子締合體,這種離子締合體可以是由兩個電荷相同的異性離子組成的離子對,也可以是由三個離子或更多個離子締合而成的離子簇團.由於離子在溶液中不停地運動,一些離子締合體存在的時間可能是短暫的.在溶液中每一個瞬間都有許多離子締合,同時又有許多締合體分解.從統計的觀點來看,溶液中總是有一定數量的離子締合體存在.締合體是靠庫侖力形成,它和靠化學鍵形成的分子是不同的.顯然電荷數大的離子在相對介電常數小的溶劑中,離子間庫侖引力較大,因而離子締合的可能性也就大些.由於締合體作為一個整體在溶液中存在和運動,所以,在一定濃度的電解質溶液中,並非每個離子都能獨立運動.對於強電解質而言,在溶液中雖然完全離子化,但並非完全解離.離子的這種締合作用顯然會影響與離子數量有關的電解質溶液的性質.
離子水化
在電解質水溶液中,除了離子之間的相互作用外,離子和水分子之間也會發生相互作用,這種作用稱為離子的水化作用,如果是泛指一般的溶劑,則稱為溶劑化作用.如圖7-1所示,離子發生水化作用時,一些極性水分子在離子周圍取向,與離子緊密結合,形成水化離子.水分子被束
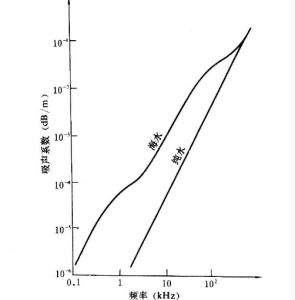 溶液吸收電解質溶液的聲吸收
溶液吸收電解質溶液的聲吸收縛在離子周圍的溶劑化層內,不能獨立移動,只能與離子一起移動,使游離的水分子數量減少,相當於離子實際濃度增大.當溶液很稀時,由於水分子數量遠遠大於離子數,幾乎所有水分子都是自由的,故水化作用對濃度的影響較小.但隨著濃度的增大,自由水分子所占比率越來越少,其影響也越來越大.
離子與水分子間的作用力在兩者之間的距離超過幾納米時,已可忽略不計,因此離子周圍存在著一個對水分子有明顯電場作用的空間.在這個空間內含有的水分子數稱為離子水化數.緊靠著離子的第一層水分子與離子結合得比較牢固.它們基本上能與離子一起移動,不受溫度的影響,這部分水化作用稱為原水化或化學水化.它所包含的水分子數目稱為原水化數.第一層以外的部分水分子也受到離子的吸引作用,使這部分水分子之間原有的結構狀態發生改變.與離子的聯繫比較鬆散的這部分水化作用,叫做二級水化或物理水化.溫度對它的影響很大,這部分水分子不與離子一起移動.測定原水化數的方法有多種,但所得結果很不一致.例如,Na+的水化數可由2到7.這是因為各種方法測出的水化數,實際上都是原水化數加上部分二級水化數,而每種方法中所包括進去的多少又各有不同.不過,在充分考慮了離子與水分子的各種相互作用能之後,可以通過統計力學方法,比較可靠地計算出離子水化數.實際上,離子水化數隻代表與離子相結合的水分子的有效數目.離子水化的一般規律是:離子半徑越小,或所帶的電荷越大,則離子表面的靜電勢能就越高,離子的水化作用也就越強,水化數也就越大.
離子電遷移
移電解質溶液中的離子,在沒有外力作用時,時刻都在進行著雜亂無章的熱運動.在一定時間間隔內,粒子在各方向上的總位移為零.但是在外力作用下,離子沿著某一方向移動的距離將比其它方向大些,遂產生了一定的淨位移.如果離子是在外電場力作用下發生的定向移動,我們稱為電遷移.離子的電遷移不但是物質的遷移,而且也是電荷的遷移,所以離子的電遷移可以在溶液中形成電流.由於正負離子沿著相反的方向遷移,所以它們的導電效果是相同的,也就是說正負離子沿著同一方嚮導電.
離子的電遷移速率除了與離子的本性(離子半徑,所帶電荷),溶液的濃度,粘度及溫度等有關外,還與電場的電勢梯度有關.在其它條件一定時,離子電遷移的速率v與電勢梯度成正比,即
v=U(7.1-1)
式中U為比例係數,稱為離子的電遷移率,其物理意義是離子在單位電勢梯度下的電遷移速率,單位是m2.V-1.s-1.離子的電遷移率是表征離子在電場中遷移的基本參數,是離子的特性.表7-1是一些離子在298.2K時無限稀釋水溶液中的電遷移率U∞,它表示的是在離子之間無相互作用時的電遷移率.從表中可見離子電遷移率很小,數量級為10-8m2.s-1.V-1,所以電解時離子的移動通常很緩慢.當電解質溶液中的電勢梯度為1000V.m-1時,離子遷移速率的數量級僅為10-5m.s-1,這比室溫下離子熱運動的速率100m.s-1要小得多.
表7-1298.2K時無限稀釋水溶液中一些離子的電遷移率
導電過程
電解質溶液的導電過程
能導電的物體稱為導體.導體分為兩類:一類是電子導體,如金屬,石墨,某些金屬氧化物(如PbO2,Fe3O4),金屬碳化物(如WC)等,它們是靠自由電子在電場作用下的定向移動而導電的.當電流通過這類導體時,除了可能產生熱量外,不發生任何化學變化.電子導體(例如金屬導線)能夠獨立地完成導電任務;另一類是離子導體,如熔融的電解質,固體電解質和以水或其它有機物為溶劑而形成的電解質溶液,它們是靠離子在電場作用下的定向移動而導電的.離子導體(例如CuSO4溶液)不能獨立完成導電任務,欲使離子導體導電,必須有電子導體與之相連線.例如,為了使電流在電解質溶液中通過,需要在溶液的兩端分別插入金屬導體,才能構成通路,於是就形成了金屬-溶液-金屬串聯的系統(構成這種系統的裝置就是電化學裝置),其中的金屬就是兩個電極.當電流通過離子導體時,除了可能產生熱量外,在兩個電極與溶液的接觸面上必然伴隨有化學反應發生和化學能與電能間的相互轉化.
下面分別討論在電池和電解池中電解質溶液的導電過程.
圖7-3(a)為一電解池,當插在HCl水溶液中的Pt片A和B,分別用導線與外電源的負極和正極接通後,在電源電場力的作用下,電源負極的電子通過導線遷移到鉑電極A上,同時鉑電極B上的電子通過導線遷移到電源正極.要想維持金屬導體電子的流動,鉑電極A必須不斷地失去電子,鉑電極B必須不斷地得到電子.由於電子不能從電極A直接進入溶液到達電極B,因此在電極A和溶液的界面處就發生了消耗電子的還原反應過程
2H++2e-=H2
在電極B和溶液的界面處就發生了產生電子的氧化反應過程
2Cl-=Cl2+2e-
同時,由於鉑電極A,B上分別帶有負電荷和正電荷,使兩電極間的溶液中存在有電場,所以兩電極間電解質溶液中的H+,Cl-就會在電場力作用下定向移動,從而形成了溶液中的電流.
圖7-1(b)為一原電池.當H2和Cl2分別衝擊插在HCl水溶液中的Pt電極A,B時,在電極A與溶液界面處,H2發生氧化反應
H2→2H++2e-
電子留在電極A上,使該電極帶上負電,H+進入溶液,使電極A附近的溶液帶上正電.同樣在電極B與溶液的界面處,Cl2發生還原反應
Cl2+2e-→2Cl-
電極B失去電子,帶上正電,Cl-進入溶液,使電極B附近的溶液帶上負電.這樣,當兩電極上的反應分別達到平衡時,兩電極間就有一定的電勢差.當外電路斷開時,兩電極上所帶電荷產生的電場(電場強度的方向由A指向B)與兩電極附近溶液所帶電荷產生的電場(電場強度的方向由B指向A)大小相等,方向相反.所以,在溶液內部,電場強度處處為零,電勢處處相等,因此離子不產生電遷移,沒有電流通過.當外電路接通時,電極B上的電子在電場的作用下,通過導線流向電極A,也就是形成了自電極A流向電極B的電流,從而使兩電極上的電荷減少,破壞了原來的平衡,導致了下面兩種現象的同時發生:一是電極與溶液之間的電場變弱,於是,H2和Cl2又在化學力作用下進行反應,來補充兩電極減少的電荷;二是兩電極上的電荷在兩極間溶液中產生的電場變弱,小於了兩電極附近溶液中的電荷在兩電極間溶液中產生的電場,因此在溶液中的電場強度不再處處為零,電勢不再處處相等,而是電極A附近的溶液中的電勢高於電極B附近溶液中的電勢,於是溶液中的H+,Cl-在電場的作用下,分別向電極B和A遷移,形成了溶液中的電流.
可見電解質溶液的導電過程,必須既有電解質溶液中離子的定向遷移過程,又有電極上物質發生化學反應的過程,兩者缺一不可,否則就不可能形成持續的電流.
為了討論問題的方便,習慣上把電化學裝置中的兩個電極按下列兩種方法命名:(1)發生氧化反應的電極叫陽極,發生還原反應的電極叫陰極;(2)電勢較高的電極稱為正極,電勢較低的電極稱為負極.在討論電解池時常使用第(1)種命名法,在討論原電池時常使用第(2)種命名法.但有時不論對電解池還是原電池,兩種命名法都用,此時要注意兩者的對應關係.
法拉第定律
1833年,法拉第在研究電解作用時,從實驗結果中歸納出一條規律:電流通過電解質溶液時,電極上發生化學反應的物質的量與通過溶液的電量成正比.後來人們稱之為法拉第定律.
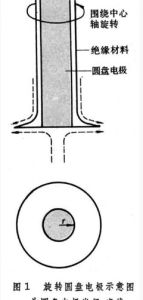 電解質溶液保持靜止不動,稱靜止電極技術
電解質溶液保持靜止不動,稱靜止電極技術根據電化學原理,很容易得到在電極上發生反應的物質的量與通過溶液的電量之間的關係式.
設電極反應計量方程式可表示為:
0=∑νBB+νee-(7.2-1)
式中B表示電極反應中的反應物或產物的化學式(分子式或離子式等),e-表示電極反應中的電子,νB和νe分別是兩者的計量係數.當B為反應物時νB取負值,當B為產物時νB取正值;對於氧化反應νe取正值,對於還原反應,νe取負值.例如,對於氧化反應H2O→O2+H++4e-,νe=4;對於還原反應Cr2O72-+14H++6e-→2Cr3++7H2O,νe=-6.
當電極反應的反應進度為ξ時,通過電極的元電荷的物質的量為
ne=|νe|ξ(7.2-2)
1mol電子所帶電量的絕對值是個常數,稱為法拉第常數,用符號F表示,定義為阿佛加德羅常數L與元電荷e-的乘積,即
F=Le=6.0221367×1023mol-1×1.60217733×10-19C=96485.309Cmol-1
在一般計算中可以近似取F=96500Cmol-1.顯然通過溶液的電量Q與ne的關係為:
Q=neF=|νe|ξF(7.2-3a)
在套用時常用z代替|νB|,並稱之為反應的電荷數(即轉移電子數),這時
Q=zξF(7.2-3b)
所以,在電極上發生反應的物質的量和質量分別為:
(7.2-4)
(7.2-5)
式(7.2-3)和式(7.2-5)均可稱為法拉第定律的數學表達式.
法拉第定律是一個從電解過程中總結出來的準確定律,但它對原電池也同樣適用.該定律不受溫度,壓力,電解質溶液的組成和濃度,電極的材料和形狀等任何因素的影響,在水溶液中,非水溶液中或熔融鹽中均可使用.
必須注意,在實際電解時,得到的所需產物的量往往比根據電量消耗按法拉第定律計算出來的量要少.為了便於說明這個問題,提出了電流效率的概念,定義如下:
電流效率=(根據法拉第定律計算所需要的電量/實際消耗的電量)×100%
或
電流效率=(實際獲得所需產物質量/根據法拉第定律計算應得所需產物質量)×100%
實際電解過程的電流效率一般都小於100%.如工業上電解精煉銅時,電流效率通常在95~97%之間,電解制鋁的電流效率約90%.引起電流效率小於100%的原因一般有以下兩種:(1)電極上有副反應發生,消耗了部分電量.例如鍍鋅時,陰極上除了有Zn2+發生還原的主反應外,還有H+發生還原的副反應.(2)所需要的產物因一部分發生次級反應(如分解,氧化,與電極物質或溶液中的物質反應等)而被消耗.例如,電解食鹽水溶液時,陽極上產生的Cl2又部分溶解在電解液中,形成次氯酸鹽和氯酸鹽.
根據法拉第定律,用電極上發生反應的物質的量可以精確計算出通過電路的電量.利用這個原理設計的測量電量的裝置稱為電量計或庫侖計.這種儀器是由電解質溶液和置於其中的兩個電極所構成.使用時,將其串聯到電路中,通電一段時間後,稱量電極上產生的物質的量,用法拉第定律求出所通過的電量.顯然在電量計中所選用的電極反應的電流效率應為100%或者是十分接近100%.最常用的是銀電量計,其次是銅電量計,氣體電量計等.
電導和電導率
1.電導和電導率
金屬的導電能力常用電阻來衡量.電阻越小,導電能力越強.電解質溶液的導電能力雖然也可以用電阻來衡量,但更習慣採用的是電導.電導即電阻的倒數.用符號G表示,
(7.4-1)
電導的SI單位是"西門子"(Siemens),簡稱"西",用S表示.顯然,導體的電導越大,導電能力越強.
因為
所以
令κ=(7.4-2)
則G=κ(7.4-3)
其中κ稱為電導率,即電阻率的倒數.SI單位是"西每米"(Sm-1).對於電解質溶液而言,式中A表示兩個相同電極中一個電極的面積,l表示兩平行電極間的距離.電導率則表示面積為1m2,相距1m的兩平行電極板之間包含的溶液的電導.電解質溶液的電導率與電解質的種類,溶液的濃度及溫度等因素有關.圖7-7是幾種電解質溶液的電導率隨濃度的變化曲線.可以看出:
(1)同溫同濃度下強酸和強鹼因能解離出H+和OH-,電導率最大,鹽類次之.弱電解質因為在溶液中不完全解離,電導率最小;
(2)不管是弱電解質還是強電解質,其電導率隨濃度的變化都是先增大,越過極值後又減小.這是因為濃度增大時參與導電的離子數目增多,使導電能力增強,隨著濃度的增大,離子間的相互作用逐漸增強,反而又使導電能力減小減弱.弱電解質的電導率隨濃度的變化不明顯,是因為濃度增大時,雖然電解質分子數增加了,但解離度卻隨之減小,溶液中離子數目變化並不大.
了解這些情況對於生產及科學研究中合適地選用電化學裝置中的電解質是有幫助的.
2.摩爾電導率
金屬導體只靠電子導電,而且導體中電子濃度很高,所以只要把導體的幾何形狀固定了,就完全能夠顯示出各種導體導電能力的大小,電導率就足以反映出不同導體在導電能力上的差別.電解質溶液則不然,它們的電荷載體是離子,各種離子的電荷數可能不同,單位體積中離子的數量(濃度)也可以不一樣,情況比較複雜.因此為了對不同電解質溶液的導電能力進行比較,除了應規定出它們的幾何形狀之外,還要對導體中離子的數量作出規定,於是提出了摩爾電導率的概念.定義如下:
把含有1mol電解質的溶液置於相距1m的兩平行電極板之間時所具有的電導,叫摩爾電導率,用符號∧m表示.若電解質溶液的物質的量濃度
 電解質溶液
電解質溶液為c(單位為molm-3),則含有1mol電解質溶液的體積Vm為1/c,單位為m3mol-1,由圖7-8可以得到
∧m=Vmκ=(7.4-4)
∧m的單位為Sm2mol-1.據式(7.4-4),又可把摩爾電導率定義為單位濃度溶液的電導率.
由於摩爾電導率涉及物質的量濃度,所以在表示電解質溶液的摩爾電導率時,應註明"摩爾"的基本單元.通常用元素符號或化學式表示.如298.15K時,
∧m(CuSO4)=14.34×10-3Sm2mol-1
∧m(CuSO4)=7.17×10-3Sm2mol-1
顯然,∧m(CuSO4)=2∧m(CuSO4)
在用摩爾電導率比較不同電解質溶液的導電能力時,除了要求溶液的溫度和濃度相同外,應使其基本單元所帶的電荷相等.例如,要比較氯化鉀和硫酸銅溶液的導電能力時,應比較同溫同濃度時∧m(KCl)和∧m(CuSO4)的大小.
圖7-9是25℃時一些電解質在水溶液中的∧m隨的變化曲線.可以看出,無論是強電解質還是弱電解質,∧m均隨濃度的減少而增大,但兩者的變化程度差別很大.
對於強電解質,因其在溶液中完全解離,所以在其物質的量固定為1mol的前提下,濃度的改變對離子的數量沒有影響,但卻影響離子之間的作用力.當濃度降低時,離子間引力減弱,離子運動速率增加,致使∧m隨濃度的減小而緩慢增加.德國化學家科爾勞施(kohlrausch)由大量實驗結果發現,濃度極稀(通常c<0.001moldm-3)的強電解質溶液的摩爾電導率與濃度的平方根有線性關係(見圖7-9中的虛線),用式子表示為:
∧m=-A(7.4-5)
式中A在一定溫度下,對給定的電解質和溶劑而言是一個常數,是直線的截距,由直線外推至與縱軸相交處得到.可見表示的是電解質溶液在無限稀釋(c→0)時的摩爾電導率,故稱為無限稀釋摩爾電導率(又稱為極限摩爾電導率).是電解質的一個特性參數,反映了電解質在離子之間沒有作用力時所具有的最大導電能力.
對於弱電解質,因其在溶液中部分解離,且解離度受濃度的影響,所以當濃度降低時,雖然溶液中電解質的數量未變,仍為1mol,但解離度卻增大了,離子的數量增多了,致使∧m隨濃度的減少而增加.當溶液很稀時,由於解離度隨濃度的減小而迅速增大,致使∧m急劇增加,∧m與c之間不存在如式(7.4-5)的簡單關係.因此弱電解質的無法用外推法求得,科爾勞施的離子獨立運動定律解決了這個問題.
3.離子獨立運動定律和離子摩爾電導率
1875年,科爾勞施在研究極稀電解質溶液時,根據大量實驗數據發現一個規律,即在無限稀釋的溶液中,所有的電解質全部解離,而且離子間一切相互作用均可忽略,每一種離子都是獨立運動的,不受其它共存離子的影響.因此電解質溶液的可以認為是正負離子摩爾電導率λ∞之和,即對於任意電解質Mν+Xν-都有下列關係式
(7.4-6)
此式稱為離子獨立運動定律,式中,分別表示正,負離子的無限稀釋摩爾電導率.顯然,如果知道了各種離子的,則無論是強電解質還是弱電解質,均可直接用此式計算.
離子的摩爾電導率可由實驗測定.下表列出了298K時無限稀釋的水溶液中一些常見離子的摩爾電導率.
常見的電解質
強電解質
強酸:HCl,HBr,HI,H2SO4,HNO3,HClO3,HClO4等.
強鹼:NaOH,KOH,Ba(OH)2,Ca(OH)2等.
絕大多數鹽:如NaCl,(NH4)2SO4,Fe(NO3)3,BaSO4等
弱電解質
弱酸:HF,HClO,H2S,H2SO3,H3PO4,H2CO3,
弱鹼:NH3·H2O,Fe(OH)3,Al(OH)3,Cu(OH)2等.
少數鹽:HgCl2,醋酸鉛等
水(極弱的電解質)
