
藥物信息
英文化學名:Clopidogrel sulfate
Cas No:135046-48-9;120202-66-6
化學名:2-(2-氯苯基)-2-(6,7-二氫噻吩並[3,2-c]吡啶-5-基)乙酸甲酯硫酸氫鹽
中文同義詞: 二硫酸氯吡格雷;二硫酸氯匹多瑞;硫酸氯吡格雷;氯吡格 雷酸氫鹽;
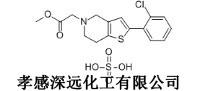 硫酸氫氯吡格雷
硫酸氫氯吡格雷外觀:白色或類白色
分子式: C15H16ClNO6S2
分子量: 405.87
標準:≧98.5%
有兩種類型
包裝規格:25kg
性質
本品為白色或類白色結晶性粉末;無臭,本品在水、甲醇、乙醇或冰醋酸中溶解,在丙酮或氯仿中極微溶解;在醋酸乙酯中幾乎不溶;在0.1mol/L鹽酸溶液中溶解。 比鏇度:+52°至+56°,儲存條件:2-8℃。
外觀:白色或類白色
分子式: C16H16ClNO6S2
分子量: 419.90
標準:≧98.5%
有兩種類型
包裝規格:25kg
適應症
氯吡格雷用於以下患者的預防動脈粥樣硬化血栓形成事件:
1、心肌梗死患者(從幾天到小於35天)、缺血性卒中患者(從7天到小於6個月)或確診外周動脈性疾病的患者。
2、急性冠脈綜合徵得患者
-非ST段抬高性急性冠脈綜合徵(包括不穩定性心絞痛或非Q波心肌梗死),包括經皮冠狀動脈介入術後置入支架的患者,與阿司匹林合用。
-用於ST段抬高性急性冠脈綜合徵患者,與阿司匹林聯合,可合併在溶栓治療中使用。
安全研究
大鼠和狒狒臨床前最常見的反應為肝臟發生變化。所服劑量為人體服用75mg/天氯吡格雷後使用劑量的25倍,這些肝臟變化是由於藥品對肝臟代謝酶影響的結果。
大鼠和狒狒服用高劑量氯吡格雷,胃耐受性差(胃炎、胃潰瘍和/或眩暈)
以每天大至77mg/kg劑量,小鼠服用78周,大鼠服用104周的氯吡格雷沒有發現致癌的證據。此劑量的血藥濃度較人類的推薦劑量(每天75mg)大25倍。
經過一系列體內的體外試驗證實氯吡格雷無致突變效果。
氯吡格雷對雌性大鼠和雄性大鼠的生育能力沒有影響,對大鼠和兔子均無致畸作用。哺乳大鼠服用氯吡格雷可輕微延緩幼兒的發育。藥代動力學研究表明氯吡格雷和/或其代謝物從乳汁中排泄,因此,不排除氯吡格雷有直接(輕微毒性)或間接(味道不好)作用。
多次口服氯吡格雷75mg以後,氯吡格雷吸收迅速,母體化合物的血漿濃度很低,一般在用藥2小時後低於定量限(0.00025mg/L)。根據尿液中氯吡格雷代謝物排泄量計算,至少有50%的藥物被吸收。
氯吡格雷主要由肝臟代謝。血中主要代謝產物是羧酸鹽衍生物,其對血小板聚集也無影響,占血漿中藥物相關化合物的85%。多次口服氯吡格雷75mg以後,血藥濃度約在1小時後達峰(30mg/l)
氯吡格雷主要由一種藥物前體,通過氧化作用形成2-氧基-氯吡格雷,然後再經過水解形成活性代謝物(一種硫醇衍生物)。氧化作用主要由細胞色素P450同功酶2B6和3A4調節,1A1、1A2和2C19也有一定的調節作用。體外分離這種活性代謝物顯示它可迅速不可逆的與血小板受體結合,從而抑制血小板聚集。但在血中未檢測到此種代謝物。
在氯吡格雷50-150mg範圍內,主要代謝物藥代動力學為線性增長(血漿濃度與劑量成正比)。
在很廣的濃度範圍內,氯吡格雷及其主要代謝物均可在體外與人體的血漿蛋白可逆性結合(分別為98%和94%)。
人體口服14C標記的氯吡格雷以後,在5天內約50%由尿液排出,約46%由糞便排出,一次和重複給藥後,血漿中主要代謝產物的消除半衰期為8小時。
每天重複服用波立維75mg後,嚴重腎損害病人(肌酐清除率5-15ml/min)和健康志願者。儘管ADP誘導血小板聚集抑制作用低於健康志願者25%,但出血時間的延長與每天服用氯吡格雷75mg的健康志願者相同。而且,所有病人的臨床耐受性良好。
健康志願者及患有肝硬化(Child-Pugh classA或B)病人單次,多劑量服用氯吡格雷,對氯吡格雷藥效學硬化病人單次,多劑量服用氯吡格雷,對氯吡格雷藥效學及藥代動力學進行研究。表明每天一次服用氯吡格雷75mg,進行10天,藥物安全,受試者對藥物耐受良好。肝硬化病人單次服藥及穩態氯吡格雷血藥濃度峰值高於健康志願者幾倍。但肝硬化組和健康志願者組間血中主要代謝物濃度,結ADP誘導血小板聚集的抑制作用和出血時間均相當。
適應症
波立維適用於有過近期發作的中風、心肌梗死和確診外周動脈疾病的患者。該藥可減少動脈粥樣硬化性事件的發生(如心肌梗死,中風和血管性死亡。)
用法用量
波立維的推薦劑量為每天 75mg,與或不與食物同服,對於老年患者不需調整劑量。
禁忌
1、對藥品或本品任一成份過敏。
2、嚴重的肝臟損傷
3、活動性病理性出血,如消化性潰瘍或顱內出血。
