環介導等溫擴增(Loop-mediated isothermal amplification,LAMP)是眾多核苷酸擴增技術(the nucleic acid amplifications tests,NATs)中的一種。自從Notomi等研究者於2000年公布該技術以來,該技術已經被廣泛地套用於生命科學領域,其中就包括對病原體感染的檢測。當研究者將該技術套用於檢測各種病原體的同時,也對該技術提出了很多的改進,使得該技術逐步完善,能夠成功地被套用於如瘧疾、錐蟲病、泰勒爾梨漿蟲病和巴貝蟲病等疾病病原體的檢測。
LAMP過程及原理
研究者稱LAMP已經被廣泛地套用於生命科學領域中各個角落的DNA或RNA的特異高效擴增。這很大程度上要歸功於它特殊的擴增原理。現通過DNA擴增過程簡單介紹其擴增原理,其擴增目的片段時依賴的是一種具有鏈置換特性的Bst DNA聚合酶(Bacillus stearothermophilus DNA polymerase)和四條能夠識別靶序列上六個特異區域的引物。靶序列的擴增反應需要在等溫條件下進行約一個小時。
 過程
過程引物的設計
通過GENBANK等途徑獲得目的片段序列。在模板兩端劃分六個區域。因為鏈的取代反應是限速步驟之一,所以目的片段的大小會影響LAMP的反應效率,一般要求目的片段小於300bp,其中包括F2和B2區。登入Eiken Chemical公司的線上軟體PrimerExplore(http://www.primerexplorer.jp/e/)設計引物。設計一對外引物F3和B3,一對內引物FIP和BIP。內引物FIP由F1c、F2(F2c的互補序列)及中間間隔區組成,BIP由B1c、 B2(B2c的互補序列)及中間間隔區組成。中間間隔區可以是-TTTT-也可以是一些特異性酶切位點。LAMP反應的開始階段四條引物都被使用,但在循環階段則只有內引物被使用。對引物設計的要求是能形成環狀結構,這也是LAMP不需要進行熱循環的關鍵點。可以這么說:LAMP反應中,引物設計是關鍵。需要注意一下幾個方面:Tm值、引物末端的穩定性、GC含量和二級結構,引物之間的距離。Tm值:利用毗鄰法計算Tm值。對於GC含量豐富或正常的模板其Tm值約在60℃~65℃,而AT含量豐富的則在55℃~60℃之間。
引物末端的穩定性:F2/B2、F3/B3、LF/LB的3’末端和F1c/B1c的5’末端作為DNA合成的起點必需有一定的穩定性,自由能應該≤-4kcal/mol。
GC含量:一般在40%-65%,而50%-60%尤其好。
二級結構:避免引物3’末端的互補及引物之間形成二聚體。
引物之間的距離:F2和B2之間的距離(LAMP擴增的區域)在120-180bp不能大於200bp,F2的5’端到F1的5’端間距(成環的區域)為40-60bp.F2和F3間距為0-20bp。
反應順序
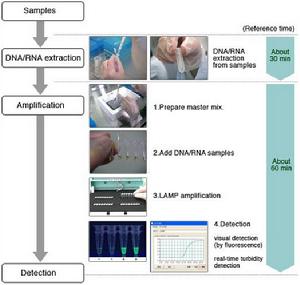
將模板、Bst DNA聚合酶、引物和其它反應試劑混合後,置於60℃-65℃的水浴鍋中,反應1h左右。
第一步:內引物FIP的F2與其模板的互補序列F2c結合,在Bst DNA聚合酶作用下,從F2的3’末端開始啟動DNA合成,合成一條以FIP為新的DNA單鏈並與模板鏈結合形成新的雙鏈DNA。而原DNA雙鏈中與模板鏈互補的非模板鏈將被取代而游離於反應液中。這種取代過程可以解釋LAMP法並不需要對雙鏈DNA進行預變性及進行溫度循環。
第二步:以F3為起始合成的新鏈與模板鏈形成雙鏈。而原合成的以FIP為起始的DNA單鏈被置換而脫離產生一單鏈DNA,其在5’末端Flc和Fl區發生自我鹼基配對,形成莖環狀結構。
第三步:引物BIP的B2與模板鏈B2c區互補配對,合成以BIP為起始的新鏈,並與模板鏈互補形成DNA雙鏈。同時,F端的環狀結構將被打開,外引物B3與模板上B3c雜交後,以其3’末端為起點也開始合成新鏈,並使以BIP為起始的DNA單鏈從模板鏈上脫離下來,形成以FIP和BIP為兩端的單鏈。因為B1C與B1互補,F1C與F1互補,兩端自然發生鹼基配對,這條游離於液體中的DNA單鏈分別在F和B末端形成兩個莖環狀結構,於是整條鏈呈現啞鈴狀結構,此結構即為LAMP的基礎結構。
第四步:形成LAMP的基礎結構後進入擴增循環。首先在啞鈴狀結構中,以Fl 3’末端為起點,以自身為模板,進行DNA合成延伸。與此同時,FIP引物F2與環上單鏈F2c雜交,啟動新一輪鏈置換反應。使以Fl的3’末端為起點合成單鏈脫離模板而解離下來形成單鏈。在解離出的單鏈核酸上也因互補結構存在而形成環狀結構。在環狀結構上存在單鏈形式B2c,BIP引物上的B2與其雜交,啟動另一輪擴增。經過相同的過程,又形成環狀結構。LAMP的終產物為莖環DNA組成的混合物,即含有若干倍莖長度莖環結構和類似花椰菜的結構。
第五步:反應結束後對擴增產物的檢測常使用焦磷酸酶沉澱檢測(濁度檢測)、螢光檢測、凝膠電泳檢測、實時檢測。
焦磷酸酶沉澱的檢測(濁度檢測):在LAMP反應過程中,dNTP析出的焦磷酸根離子與反應溶液中的Mg2+結合,產生副產物焦磷酸酶白色沉澱,研究者發現LAMP反應中焦磷酸鎂沉澱的形成與所產生的DNA量之間的關係,發現兩者生成量之間呈線性關係,並且焦磷酸鎂沉澱在400 nm處有吸收峰,從而進行LAMP的定量檢測。
螢光檢測:LAMP有極高的擴增效率,可在一小時內將靶序列擴增至109~l010倍,所以當反應液中加入核酸染料SYBR Green I後,在紫外燈或日光下通過肉眼即可進行判定,如果含有擴增產物,反應混合物變綠;反之,則保持SYBR Green I的橙色不變;同時,也可以進行LAMP的實時定量檢測。研究者進一步改進方法,使其適用於多重檢測。同時加入特異的帶不同螢光顏色的探針,探針與特異的模板結合,反應結束後加入陽離子聚合物PEI(只與高分子量的物質結合形成沉澱複合物),與LAMP擴增產物形成沉澱,根據擴增產物上結合的特異性探針在紫外光下發射出的顏色,肉眼即可判斷結果。因為探針的存在,排除了非特異性擴增的可能。
LAMP的優勢
核酸擴增技術在生命科學領域是非常重要的一種工具,例如疾病診斷、基因功能特性的研究等。除了傳統的PCR方法,現在研究者已經開發出了很多其他的核酸擴增方法,比如NASBA(nucleic acid sequence-based amplification)、3SR(self-sustained sequence replication)、SDA(strand displacement amplification)等。在擴增循環方面,它們有各自的創新點。相比於常規PCR利用高溫使雙螺鏇解鏈變性成單鏈進行熱循環,NASBA和3SR則使用一系列轉錄和反轉錄過程來循環以避免高溫變性作用,SDA則使用限制性內切酶和修飾過的模板來循環擴增。雖然它們的敏感性都很高,可以檢測並擴增小於10個拷貝的核酸樣本,但是它們還有各自需要克服的缺點。技術要求、材料儀器要求、技術本身特異性缺陷等方面嚴重束縛了這些技術的推廣套用。LAMP則在這些方面有所突破。
LAMP有比較高的特異性和抗干擾能力,只有當2對引物與目的片段的六個區域都匹配上時才能進行擴增。類似於巢式PCR使用多對引物來提高擴增的特異性。非目的片段對LAMP反應的干擾比較小,這方面比常規的PCR要強。LAMP的反應體系比較穩定可靠,在室溫下放置2周后仍然穩定並且對於樣品中原有或污染的無關、干擾片段仍然不敏感,而其他NATs則無法做到這一點。研究者利用LAMP檢測腦脊髓液或血液樣本中的布氏錐蟲,沒有出現使用常規PCR時出現的組織或血液樣本中的抑制劑干擾反應的現象。同時LAMP的敏感性也比較高,可以以單拷貝的基因為模板進行擴增。有研究者報導使用LAMP檢測弓形蟲速殖子的最低濃度為2~3個/ml,但也有研究者報導過更低的拷貝濃度。
LAMP 反應的過程簡單快速而且高效。能夠在1h內將單拷貝的基因模板擴增到109個拷貝,而且這一過程是在60-70℃的恆溫下進行的。這就擺脫了昂貴儀器的束縛。另外LAMP結果的檢測也無需儀器。這些在一定程度上降低了實驗的操作成本。只要引物設計正確,並且完善各種反應條件之後,LAMP對於樣品處理、操作技術和儀器設備的要求都比較低,在野外工作時也能達到。另外正如上文所述LAMP 的抗干擾能力較強,使用的樣品可以是未經提純處理的,所以LAMP為野外實地開展檢測工作提供了很好的技術支持。
LAMP的改進與深化
隨著LAMP的優點逐漸為研究者所熟知,它在越來越多的領域中被使用,包括病毒病原體的檢測、細菌病原體的檢測、真菌病原體的檢測、寄生蟲的檢測、腫瘤的檢測等。當然,不同需求的研究者在使用LAMP的時候,針對各自的研究需要對LAMP進行了不同程度的改進提高及延伸。
環狀引物
通過增加2條環狀引物,使LAMP的反應時間縮短近一半,提高了檢測效率。環狀引物結合的區域在F2-F1或B2-B1,以Fl到F2的方向或是B1到B2的方向結合。當反應時,所有的莖環區DNA序列或與內引物雜交,或者與環狀引物雜交,從而加快了反應速度。
RT-LAMP
LAMP也同樣適用於RNA模板,在反轉錄酶和DNA聚合酶的共同作用下,實現RNA的一步擴增。研究者利用RT-LAMP檢測前列腺癌特異抗原(PSA),將一個表達PSA的LNCaP細胞與1 000 000個不表達PSA的K562細胞混合,提取RNA,RT-LAMP也能夠檢測得到。
原位LAMP
將LAMP技術和原位雜交技術相結合。研究者想要檢測攜帶一種編碼毒素的基因stx2的大腸桿菌O157:H7。利用細胞原位固定法,用不同螢光抗體標記大腸桿菌與無stx2特異基因的細菌混合物,從而區別出攜帶特異性基因的大腸桿菌。與原位PCR相比,溫和的滲透性及低的等溫條件使得原位LAMP對細胞的損傷減小,準確性提高。
分離單鏈DNA
LAMP的產物可以用於後續試驗,如雜交試驗,但是單鏈DNA的雜交效率要明顯高於雙鏈DNA,因而要將莖-環狀產物適當處理,利於後續反應。研究者從LAMP的產物中分離出單鏈的靶序列,用TSPR I酶消化LAMP的產物,再利用一特殊的引物在斷裂處和3′端雜交並延伸產生一條特異性DNA,利於進行雜交檢測,如DNA微列陣技術。反應中所用的DNA聚合酶仍為Bst DNA聚合酶,因其具有鏈置換活性,可置換出單鏈DNA。因為酶的最適溫度都為65℃,所以仍是等溫反應。
多重LAMP
有研究者利用多重LAMP檢測牛巴貝蟲屬寄生蟲,分別設計B.bovis和B.bige的引物,檢測靈敏度分別是傳統PCR方法檢測B.bovis和B.bige的103和105倍。
LAMP在弓形蟲檢測方面的進展及前景
檢測弓形蟲的方法有很多,其中血清學方法間接免疫螢光抗體試驗(indirect fluorescent anti-body test,IFAT)和酶聯免疫吸附試驗(enzyme-linked immunosorbent assays,ELISA)可用於檢測血清中弓形蟲的抗原抗體,但是血清學方法無法判斷感染的時間,特異性和敏感性也不太好。另外也可使用免疫組化和PCR(polymerase chain reaction)等病原學方法檢測弓形蟲,但考慮到蟲體體積小,常規的病原學檢測容易漏診。近年來修飾凝集試驗(modified agglutination test,MAT)、免疫色譜試驗(immunochromatographic test,ICT)和實時PCR(real-time PCR)也開始在弓形蟲檢測方面有所套用。PCR、免疫色譜試驗等方法無法擺脫高昂的儀器設備。相比之下,LAMP在儀器方面的低要求完全滿足了貧困地區對病原體快速檢測的需求,因為全過程只需要一個恆定的溫度就可以了,一般的水浴鍋都可以滿足這一要求,而且實驗的結果可肉眼直接觀察。這些同時也證明了LAMP具有野外使用的潛力。
對於很多基因來說它們在生物細胞內的拷貝數是很少的,如果要對這些基因進行進一步的研究,就必須要對目的片段進行人工擴增。宿主感染弓形蟲後多表現為隱性感染,體內蟲體數量少,在使用PCR方法進行弓形蟲檢測時,一般是先從樣本中提取基因組,將該基因組作為模板,使用弓形蟲特異性的引物進行擴增,通過電泳檢測目的片段來判斷樣品是否含有弓形蟲。
研究者已經利用弓形蟲速殖子表面抗原基因SAG1、蟲卵表面蛋白基因TgOWP和B1構建了相關檢測體系,並且發現這些檢測體系相比於常規的PCR方法,具有特異性,敏感性、效率、速度和操作等方面的優勢。其中使用純化模板時LAMP的敏感性要比常規PCR高1000倍左右,使用組織樣本作為模板時差距變成了1000 000倍左右,但是LAMP也需要使用環狀的引物才能達到比較好的檢測效果。依賴基因TgOWP和B1的檢測體系中,LAMP平均能夠檢測到的最低稀釋度為0.1個速殖子。隨著被檢測樣品的複雜化越來越大,LAMP相比於常規PCR的優勢逐漸擴大。有研究者通過檢測水樣中的弓形蟲蟲卵來比較巢式PCR(nested polymerase chain reaction),免疫螢光(immunofluorescence test,IFT)與LAMP。使用純化的樣品時LAMP的檢出率是100% 而巢式PCR只有53.8%;使用未處理的樣品時LAMP檢出率為48%,巢式PCR只有13.5%,而IFT為0%。
弓形蟲的蟲株比較複雜,他們都具有較多的蛋白抗原,以及很多保守的基因序列,依靠這些保守序列建立LAMP檢測體系,我們就可以快速地對弓形蟲進行檢測。其中篩選出合適的序列很重要,要考慮序列的保守性。同時還要完善各種實驗條件使反應能夠具有好的重複性以及抗外界干擾能力。
