
微區成分分析
微區成分分析技術在物理學、化學、地學、材料科學、生命科學等領域內,得到了廣泛的套用。電子束微區成分分析一般在透射電子顯微鏡、掃描電子顯微鏡或專門的掃描透射電鏡(見電子顯微鏡)內進行。早期的電子探測束X射線微分析儀發展到目前,和具有成分分析功能的掃描電鏡日益接近,可以歸併為一類。微區成分分析的儀器主要有 X射線譜儀和電子能量損失譜儀。
X射線譜儀 有X射線波譜法和X射線能譜法兩類。
X射線波譜法 這是50年代開始發展起來的方法,目前主要在掃描電鏡中套用。利用標識 X射線進行成分分析的依據是莫塞萊定律:標識X射線的頻率的平方根近似和原子序數有線性關係。測定標識 X射線波長(或頻率)的原理是布喇格定律(見X射線衍射)。X射線波譜儀中的分光晶體(如 LiF單晶等)按不同的布喇格角將不同波長的X射線光子反射到正比計數器中進行計數,得到X射線波譜。根據標識 X射線峰的位置確定試樣中存在何種元素,根據峰的高度確定此種元素的成分。
X射線能譜法 這是 70年代發展起來的方法,目前不僅在掃描電鏡,而且在透射電鏡中得到廣泛的套用。X 射線能譜儀主要由鋰漂移矽固體探測器、放大器和多道脈衝高度分析器組成。入射 X射線光子在極短的時間內在鋰漂移矽中激發出許多電子-空穴對,電子-空穴對的數目和入射光子能量成正比,輸出的脈衝信號高度也和光子能量成正比。多道分析器按脈衝高度(即光子能量)在相應的能量通道中進行計數,得到X射線能譜。根據標識X射線峰的位置和峰的面積確定某種元素的含量。
兩種X射線譜儀各有優缺點。波譜儀的優點是:①波長解析度高,標識 X射線峰的寬度比能譜儀低一個量級;②元素含量的探測限可達0.01重量百分比,比能譜儀低一個量級;③可以探測原子序數≥5的所有元素,而能譜儀一般只能探測原子序數≥11的所有元素。能譜儀的優點是:①能同時探測試樣中所有元素的標識X射線,而波譜儀一次只能測定一個標識X射線;②探測效率高,這是由於矽探測器可以放到試樣附近,收集更多的標識X射線,而波譜儀的探測效率要低1~2個量級;③探測器結構緊湊、體積小、價格低,而波譜儀則相反。總起來看,能譜儀優點更多、發展得更快,特別是它能裝配到透射電鏡上對薄試樣進行分析,使透射電鏡發展成為分析電鏡(見電子顯微鏡)。
微區成分 X射線譜分析的空間解析度由電子束激發X 射線的範圍決定。幾十千電子伏能量的電子束在厚試樣中激發X射線的範圍約1μm3。對薄試樣來說,電子束激發 X射線的範圍大體上是電子束的面積乘上試樣的厚度。如果束直徑為10nm、試樣厚100nm,則薄試樣中X射線激發範圍比厚試樣小 5個數量級。在這樣小的微區中產生的X射線光子數極少,需要用探測效率高的X射線能譜儀進行探測。
X 射線譜定量分析方法有兩種:有標樣法和無標樣法。有標樣法要求在同樣實驗條件下,分別測定已知成分標樣和未知成分試樣同一標識譜的強度,再根據強度比確定試樣中某一元素的含量。在厚試樣中必須考慮標樣和試樣原子序數(或平均原子序數)的不同、對標識譜出射過程吸收的不同和X射線引起的次級X射線螢光的不同後進行修正(即所謂的ZAF修正),才能從標識X射線強度比得到準確度較高(誤差<10%)的元素含量。薄試樣中上述三種修正在許多場合下可以忽略。無標樣法利用 X射線物理計算出來的強度代替標樣上測得的強度或直接計算出試樣成分和標識譜強度的關係,再根據試樣上測得的強度確定試樣中元素的成分。無標樣法節省了許多測試標樣的時間,但準確度略差。
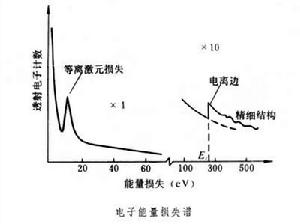
電子能量損失譜 是利用磁稜鏡(或其他電子能量分析器)獲得的透過薄試樣的電子按能量分布的曲線,一般100keV量級電子透過試樣後損失的能量較小,因此用能量損失為橫坐標表示透射電子的能量分布,得到電子能量損失譜。透射電子在磁稜鏡的磁場中發生偏轉,給定磁場後只有一種能量的電子才能通過磁稜鏡出口進入電子探測器,順序改變磁場後即可得到透射電子的能量損失譜。
由圖可見,電子能量損失譜可分為:等離(子體)激元損失峰、電離邊、擴展電離邊精細結構等組成部分。電離邊的能量損失等於某一內層 j電子的電離能Ej。利用Ej以上超出背底(圖中虛線)的電離損失譜可進行微區成分分析。
 微區成分分析
微區成分分析