
概述
嗜血細胞綜合症人體血液中的吞噬細胞是一把雙刃劍,一方面像清潔工一樣吞噬入侵的細菌和機體的衰老細胞,
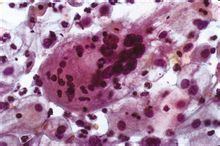 巨噬細胞
巨噬細胞綜述
噬血細胞綜合症不是遺傳性疾病。這種疾病是一種與急性病毒感染有關的良性噬血組織細胞增生,是血液內科一種少見的疾病。病因可能是感染、藥物或腫瘤引起的。多發於兒童,其特點為單核-巨噬細胞增生活躍,並有明顯的吞噬紅細胞現象。患者多有明顯高熱,肝、脾和淋巴結腫大,患者有貧血現象,白細胞明顯減少,分類可見淋巴細胞明顯增高,易見異淋。血小板常減低。簡單來說就是由於感染病毒,或是因為用藥不當,以及體內有腫瘤(對於小孩來說腫瘤幾率低)等,很可能是由於感冒或者是小型傷口感染。嬰幼兒感染後由於自身抵抗能力弱。調節不當。致使單核-巨噬細胞增生(一種機體重要的免疫細胞清除病毒腫瘤細胞等)而且對自身血細胞發生攻擊並消除。使得嬰兒貧血導致死亡。較大的有血液專科的醫院均可以治療,但是目前無哪一家醫院能保證百分之百治癒該病。治療費用較高。綜合症分類
一類為原發性或家族性,遺傳性占主要因素。
二類
為繼發性,後者可由感染及腫瘤所致。原發性HPS,或稱家族性HPS,為常染色體隱性遺傳病,其發病和病情加劇常與感染有關;繼發性HPS分為感染相關性HPS(infection-associatedhemophagocyticsyndrome,IAHS),此型多與病毒感染有關,由病毒引起者稱病毒相關性HPS(virus-associatedhemophagocyticsyndrome,VAHS);由腫瘤引起者稱腫瘤相關性HPS(malignancy-associatedhemophagnocyticsyndrome,MAHS)。
流行病學
以兒童多見,男性多於女性。兒童原發性HLH(FHL)的年發病率約為0.12/1O萬。在日本和亞洲國家發病率較高。本病來勢兇險,東方患者的死亡率約為45%。病因和發病機制
HPS可以看作細胞因子病(cytokine disease),或巨噬細胞激活綜合徵。作為免疫應答的反應性T細胞(Th1和Tc)和單核吞噬細胞過度分泌淋巴、單核因子〔巨噬細胞增生的誘導因子(PIF)〕激活巨噬細胞。惡性細胞亦可直接刺激組織細胞,或由腫瘤細胞產生釋放細胞因子(如γ-干擾素),誘發臨床綜合徵,稱之為副新生物綜合徵(para-neoplastic syndrome)。高細胞因子血症作為血細胞減少和器官衰竭的中間機制。CD+4T細胞分泌誘導巨噬細胞增生的因子(PIF)為HPS的始動因素。IFNγ和TNF-α引起骨髓造血抑制,IFN-γ、TNFα和IL-1導致發熱、肝功能異常、高脂血症及凝血障礙。可溶性白介素2受體(sIL-2R)的過度增高結合IL-2可作為抑制正常免疫反應的“阻斷因子”導致繼發性免疫缺陷狀態。目前認為HPS患者血細胞減少有多種因素參與:①噬血細胞增多,加速血細胞的破壞;②血清中存在造血祖細胞增殖的抑制性物質,骨髓內粒系和紅系前體細胞和巨核細胞進行性減少,歸因於抑制性單核因子和淋巴因子的產生,諸如γ-干擾素、腫瘤壞死因子(TNF)和白介素-1以及造血生長抑制因子的產生。
HPS的發病機制:①存在免疫調節障礙或免疫失衡;②淋巴和單核因子持續產生,作為免疫應答的反應性T細胞分泌淋巴因子可活化巨噬細胞,尤其如γ-干擾素不僅能抑制造血,而且亦能活化巨噬細胞,淋巴因子GM-CSF亦激活巨噬細胞;③遺傳因素影響機體對感染的反應方式,如家族性噬紅細胞性淋巴組織細胞增生症和X聯淋巴增殖綜合徵的兒童可發生類似的血液學異常;④存在單克隆性T細胞增殖,在EB病毒相關噬血細胞綜合徵(EBV-AHS)的患者採用PCR法檢測10/11例呈TCRγ鏈重排,亦有報導TCRβ基因的單克隆性重排,顯示EB病毒感染T細胞引起單克隆增殖的可能,或許是末梢T細胞“腫瘤”的一種特殊類型。EBV-AHS患者EBV整合入宿主T細胞染色體基因組造成單克隆T細胞增生(從良性到新生物前期或明顯的惡性增殖)伴異常的T細胞。為何異常的T細胞反應導致組織巨噬細胞的吞噬行為改變,可能由T細胞過度分泌的淋巴因子所介導。
噬血細胞綜合徵按其病因,除兒童期發病的家族性HPS(FHL)之外,可分為原發性(原因不明)或繼發性,繼發性HPS常見病因為感染、藥物、紅斑狼瘡、實體瘤和血液系腫瘤及免疫缺陷等,故一旦HPS診斷確定,應嚴格探究潛在疾患。感染相關的HPS(IAHS)中多見病毒(尤其是EB病毒)和細菌感染,血液腫瘤多見於惡性淋巴瘤,已報導可引起HPS的惡性淋巴瘤有外周T細胞淋巴瘤,NK細胞淋巴瘤,血管中心型淋巴瘤,成人鼻T細胞淋巴瘤,大細胞性淋巴瘤(T-和B-細胞型),Ki-1陽性大細胞淋巴瘤(即間變性大細胞淋巴瘤),免疫母細胞淋巴結病樣T細胞淋巴瘤和進展性NK細胞白血病。
潛在性疾患有:
感染①病毒(EB病毒、皰疹病毒、巨細胞病毒、登革熱病毒、水痘病毒、帶狀皰疹病毒、B肝病毒、副流感病毒Ⅲ等);②細菌(傷寒桿菌、不動桿菌、大腸桿菌、布氏桿菌、結核桿菌、金黃色葡萄球菌、β-溶血性鏈球菌、草綠色鏈球菌、糞鏈球菌、肺炎球菌);③支原體;④真菌(念珠菌、隱球菌、莢膜組織胞漿菌);⑤立克次體(恙蟲病、Q熱等);⑥原蟲(利什曼原蟲、瘧原蟲);新生物骨髓增生異常綜合徵(MDS)、急性非淋巴細胞白血病、T或B細胞淋巴瘤、“組織細胞”淋巴瘤、慢性淋巴細胞白血病、霍奇金病、多發性骨髓瘤、毛細胞白血病、轉移性癌腫、胃癌、惡性畸胎瘤等。
免疫介導性疾病系統性紅斑狼瘡、脂膜炎、類風濕性關節炎、結節病、炎性腸病等。
免疫缺陷狀態免疫抑制劑和(或)細胞毒藥物治療、脾切除、愛滋病、X-聯淋巴增生綜合徵。
其他壞死性淋巴結炎、成人Still病、慢性腎衰、腎移植後、飲酒過量等。
噬血細胞綜合徵通常是某種疾病的中間狀態,在某一階段該疾病有噬血細胞綜合徵的表現。
病理
骨髓塗片中出現體積較大的噬血組織細胞,吞噬物為形態完整的白細胞,有核紅細胞,成熟紅細胞及血小板,亦可為不完整的細胞及細胞碎片等,鹼性磷酸酶染色陽性率及積分正常或增高。吞噬性組織細胞增多累及骨髓、淋巴結竇狀隙和髓索、脾紅髓、肝血竇和門脈區,偶可浸潤其他器官,如肺、心、腎上腺、中樞神經系統、腎、子宮和胃。發病機理
嗜血細胞綜合徵可分為原發生和反應性,潛在疾患可為感染、腫瘤、免疫介導性疾病等,由於噬血細胞增多,加速了血細胞的破壞。因為老年人的體質比較弱,免疫力較差所以更容易會發生感染,這個病一般是不會傳染的,如果是感染因素,要儘快去除感染因素,原發病治療好後多可自愈病理特徵
良性組織細胞增加並伴嗜血現象,多見於淋巴結的淋巴竇和髓索、肝竇、門靜脈、脾臟的紅髓和骨髓。在急性期,該病與白血病、惡性組織細胞增生症、傳染性單核細胞增生症等病相似,且並非所有病例第一次骨穿即能發現嗜血細胞,有時需多部位穿刺才能確診。骨髓多增生活躍,粒系統所占比例降低,中性粒細胞可見毒性變。幼紅系統增生多正常,淋巴系統比例亦未見明顯改變,可見異淋。單核巨噬系統增生活躍,常>10%,巨噬細胞大小為20~40微米,或更大,胞漿豐富,吞噬多個成熟紅細胞,或幼紅細胞或血小板等。巨核細胞大致正常。診斷標準
1、發熱:發熱超過1周,熱峰>38.5'C2、肝脾腫大:肝脾大伴全血細胞減少,累計>=2個細胞系
3、血細胞減少(外周血二或血三系細胞減少),其中血紅蛋白<90g/L,血小板<100x109/L,中性粒細胞<1.Ox109/L
4、高甘油三酯血症和/或低纖維蛋白原血症
5、骨髓、脾或淋巴結可見噬血細胞但無惡性表現。
感染性嗜血細胞綜合症是一種與急性病毒感染有關的良性噬血組織細胞增生症,多發於兒童,其特點為單核-巨噬細胞增生活躍,並有明顯的吞噬紅細胞現象。患者多有明顯高熱,肝、脾和淋巴結腫大,原發病治療好後多可自愈。患者有貧血現象,白細胞明顯減少,分類可見淋巴細胞明顯增高,易見異淋。血小板常減低。
骨髓多增生活躍,粒系統所占比例降低,中性粒細胞可見毒性變。幼紅系統增生多正常,淋巴系統比例亦未見明顯改變,可見異淋。單核巨噬系統增生活躍,常>10%,巨噬細胞大小為20~40微米,或更大,胞漿豐富,吞噬多個成熟紅細胞,或幼紅細胞或血小板等。巨核細胞大致正常。
鑑別方法
1.結合臨床表現和實驗室檢查結果,我更傾向於嗜血細胞綜合徵,可能為病毒感染相關性,仔細看看血片能否見到異形淋巴細胞。如果比例在臨界值,你就保守一點,報“不除外嗜血細胞綜合徵,請結合臨床考慮”。2.關於嗜血細胞綜合徵骨髓象中嗜血細胞比例的問題,我認為不能對2%認得太死,因為嗜血細胞體積較大,推片時易於推至片尾和邊緣,所以按常規體尾交界處分片法,可能比例較低,但片尾和邊緣可能較多,我認為應該縱觀全片,只要片尾和邊緣嗜血細胞明顯增多,甚至成堆分布,即使體尾交界處比例達不到2%,依然可以診斷。
併發症
出血、感染、多臟器功能衰竭和DIC。繼發性噬血細胞綜合徵
繼發性HPS針對病因進行相應治療。對HPS或高細胞因子血症的治療對策為:①類固醇療法或大劑量甲基強的松龍衝擊;
②靜脈滴注大劑量丙種球蛋白(多用於VAHS);
③抑制T細胞活化的特異性抑制劑環孢菌素A或聯用G-CSF治療VAHS,或抗胸腺細胞球蛋白;
④直接拮抗細胞因子的抗TNF抗體和IL-1受體拮抗劑;
⑤為抑制或減少淋巴因子的供應源可採用化療。包括CHOP、CHOPE方案或緩慢靜滴長春新鹼。屢已報導套用依託泊甙(VP16)治療原因不明的重症HPS、EBV-AHS或LAHS奏效。預後分析表明,對於不易與MH鑑別的HPS患者啟用化療是必需的;
⑥骨髓掃蕩性(根治性)治療和異基因骨髓移植(allo-BMT)或外周血幹細胞移植治療FHL或耐化療的LAHS或EBV-AHS病例,優於常規化療和免疫抑制治療。
腫瘤相關性噬血細胞綜合徵治療方案決定於疾病的類型,如HPS發生於治療前的免疫缺陷患者,則治療主要是抗感染及抗腫瘤;如果HPS發生於化療後,而腫瘤已緩解則應停止抗腫瘤治療,同時抗感染,加用腎上腺皮質激素及VP16;對進展迅速的MAHS則應針對細胞因子所致的損害進行治療,可用前述HLH94方案。
預後
Kaito等報導住院期間的危險因素為總膽紅質增加(P=0.0001)、血小板進行性減少(P=0.0015)、貧血(P=0.002)和血清ALP增高(P=0.005)。與死亡相關的危險因素為年齡>30歲,存在彌散性血管內凝血(DIC)(FDP>10μg/ml),鐵蛋白(>500ng/ml)和β2微球蛋白(>3.0μg/ml)增高,貧血(Hb<100g/L)伴血小板減少(<100×109/L)和黃疸。此外,無淋巴結腫大與預後不良顯著相關(P=0.022)。作者指出具有HPS危險因素的患者應該積極化療和支持治療。Imushuku,等認為高細胞因子血症,如IFN-γ增高與HPS的嚴重度相關。Ishii等強調血清TNF濃度>50pg/ml生存期最短。Ohga指出血清IFN-γ值為反映HPS病情發展的最敏銳的指標。Fujiwara等檢測小兒HPS32例,其中IFN-γ值29/32例、IL-6值31/32例、sIL-2R值30/32例增高,認為這三項的異常增高是HPS的預後指標。由單核/巨噬細胞由來的TNF-α、IL-1β值增高,分別見於9/16例,5/27例。HPS預後不良,取決於潛在疾患的嚴重性及細胞因子暴(cytokinestorm)的強度,約有半數病例死亡。呈暴發性經過者病情急劇惡化,4周內死亡。生存者1~2周血細胞數恢復,肝功能恢復需較長時間(3~4周)。Kaito等報導HPS患者34例中14例生存,20例(58.5%)死亡,20例死亡者中13例(65%)為原因不明的HPS患者。Wong等報導40例東方人群反應性(R)HS,其中18例(45%)死於RHS或潛在疾病的併發症。Cline報導HPS23例中57%死亡。血液惡性疾患患者中,合併HPS和不合併HPS組平均生存期分別為7個月、48個月,呈顯著性差異。T/NK-LAHS患者預後絕對不良。主要死亡原因為出血、感染、多臟器功能衰竭和DIC。Elizabeth等報導52例伴VAHS的致死性傳染性單核細胞增多症(IM),認為繼發於EB-VAHS的骨髓損害(組織細胞吞噬血細胞和骨髓壞死)在導致IM死亡中起主要作用,死因為繼發感染和出血。
