成份
主要組成成份:佐米曲普坦
化學名稱:(s)-4-[[3-[2-(二甲氨基)乙基]-1H-吲哚-5-基]甲基]-2-噁唑烷酮。
化學結構式:
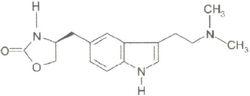 佐米曲普坦片
佐米曲普坦片分子式:CHNO
分子量:287.36
性狀
本品為白色或類白色片。
適應症
適用於成人伴或不伴先兆症狀的偏頭痛的急性治療。
規格
2.5mg
用法用量
治療偏頭痛發作的推薦劑量為2.5mg(1片)。如果24小時內症狀持續或復發,再次服藥仍有效。如需二次服藥,時間應與首次服藥時間最少相隔2小時。服用本品2.5mg(1片),頭痛減輕不滿意者,在隨後的發作中,可服用5mg(2片)。通常服藥1小時內效果最明顯。偏頭痛發作期間無論何時服用本藥。都同樣有效,建議發病後儘早服用。反覆發作時,建議24小時內服用總量不超過15mg(6片)。本品不作為偏頭痛的預防性藥物。腎損害患者使用本品無需調整劑量。
不良反應
本藥耐受性好。不良反應很輕微/緩和、短暫,且不需治療亦能自行緩解。可能的不良反應多出現在服藥後4小時,繼續用藥未見增多。最常見的不良反應包括:偶見噁心、頭暈、嗜睡、溫熱感、無力、口乾。感覺異常或感覺障礙已見報導。咽喉部、頸部、四肢及胸部可能出現沉重感、緊縮感和壓迫感(心電圖上沒有缺血改變的證據),還可出現肌痛、肌肉無力。
禁忌
禁用於對本品任何成份過敏的患者。血壓未經控制的病人不應使用。
注意事項
本藥僅套用於已診斷明確的偏頭痛患者。要注意排除其它嚴重潛在性神經科疾病。
尚無偏癱性或基底動脈性偏頭痛患者使用本品的資料,不推薦使用。
症狀性帕金森氏綜合症或患者與其它心臟旁路傳導有關的心律失常者不應使用本品。
此類化合物(5HTID激動劑)與冠狀動脈的痙攣有關,因此,臨床試驗中未包括缺血性心臟病患者,故此類患者不推薦使用本品。由於還可能存在一些未被識別的冠狀動脈疾病患者,所以建議開始使用5HTID激動劑,治療前先做心血管的檢查。與使用其他5—HTID激動劑類似,服用佐米曲普坦後,心前區可出現非典型心絞痛的感覺;但是臨床試驗中,此類症狀與心律失常或心電圖上顯示的缺血改變無關。
目前尚無肝損害者使用本品的臨床或藥代動力學的經驗,不推薦使用。
對駕駛及機械操縱能力無明顯損害,本藥20mg時,患者在精神運動測試中,操縱項目未見明顯的損害。使用本品不會損害患者駕駛及機械操縱的能力,但仍要考慮到本藥可能引起嗜睡。
孕婦及哺乳期婦女用藥
目前還沒有妊娠婦女使用本品的研究。應該只有在對母親的益處大於對胎兒的潛在性危險的情況下,才考慮使用本藥。哺乳動物試驗顯示佐米曲普坦可進入乳汁,尚無人類使用的資料。因此,哺乳期婦女慎用。
兒童用藥
本品用於兒童患者的安全性和有效性尚未確定。
老年用藥
用於65歲以上患者的安全性和有效性尚無系統的評價。
藥物相互作用
1、沒有證據表明,使用偏頭痛預防性藥物(例如β—受體阻滯劑、口服二氫麥角胺、苯噻啶)對本藥的療效有任何影響。急性對症治療,如使用撲熱息痛、滅吐靈及麥角胺不影響本品的藥代動力學及耐受力。
2、在健康個體中,沒有見到本品與麥角胺有藥代動力學的相互作用。本藥與麥角胺/咖啡因合用耐受性良好,與單獨套用本藥相比,不良反應沒有增加,血壓也無改變。
3、使用本藥治療12小時內應避免使用其他5HTID激動劑。使用嗎氯貝胺(一種特殊的單胺氧化酶—A抑制劑)後,佐米曲普坦的曲線下面積有少量增加(26%),活性代謝物的曲線下面積有三倍增加。因而對於使用單胺氧化酶—A抑制劑的患者,建議24小時內服用本品的最大量為7.5mg。
4、司來吉蘭(一種單胺氧化酶—β抑制劑)和氯西汀(一種選擇性5—羥色胺再攝取抑制劑)對佐米曲普坦的藥代動力學參數沒有影響。健康老人中,佐米曲普坦的藥代動力學與健康青年志願者是相似的。
5、與西咪替丁、口服避孕藥合用時,也可使本品的血藥濃度增加。與心得安合用可延緩本品的代謝。
藥物過量
志願者單次服用本品50mg時,常常體驗到鎮靜作用。
佐米曲普坦片的清除半衰期達2.5~3小時(見藥代動力學部分);因而,對服用過量的患者,如症狀和體徵存在,應持續監測15小時以上或直至症狀/體徵消失。
對佐米曲普坦沒有特殊的解毒藥。嚴重中毒病例,建議採取監護措施,包括建立和保持患者呼吸道的通暢,保證足夠的氧氣攝入及通氣,對心血管系統進行監測和支持。
血液透析或腹膜透析對佐米曲普坦血清濃度的影響尚不清楚。
藥理毒理
藥理作用
佐米曲普坦是一種選擇性5—HTIB/ID受體激動劑。通過激動顱內血管(包括動靜脈吻合處)和三叉神經系統交感神經上的5-HTIB/ID受體,引起顱內血管收縮並抑制前炎症神經肽的釋放。
毒理研究
遺傳毒性:
Ames試驗中,在代謝活化劑存在時,5株傷寒沙門氏菌中2株顯示有致突變作用。體外哺乳動物細胞突變試驗(CHO/HGPRT)結果為陰性。體內、外人淋巴細胞試驗,無論是否有代謝活化劑存在,佐米曲普坦均顯示致染色體裂變作用;小鼠體內微核試驗未見該作用。在非程式化DNA合成試驗中也未顯示有遺傳毒性。
生殖毒性:
雄、雌性大鼠交配前到著床期間給予佐米曲普坦,劑量達400mg/kg/日(該劑量下的暴露量約為人最大推薦劑量10mg/日下的3000倍),未顯示對生育力的損害。
大鼠和家兔的生殖毒性研究中,懷孕動物口服給予佐米曲普坦可導致胚胎死亡和胎仔異常。器官形成期孕鼠口服佐米曲普坦100、400和1200 mg/kg/日(母體血漿暴露量分別約為人每日最大推薦劑量10mg下的280、1100和5000倍),導致劑量相關的胚胎死亡率增加,高劑量時具有統計學意義。高劑量產生母體毒性,表現為妊娠期間母體體重增長減少。在一項相似的家兔試驗中,母體毒性劑量10和30 mg/kg/日(母體血漿暴露量相當於人接受最大日推薦劑量10mg暴露量的11和42倍)時,胚胎死亡率增加。30 mg/kg/日劑量時,觀察到胎仔畸形(胸骨融合、肋骨異常)和變異(主要血管變異、肋骨骨化方式不規則)發生率增加。3 mg/kg/日為無作用劑量(相當於10mg劑量時人的暴露量)。雌性大鼠妊娠、分娩和哺乳期給予佐米曲普坦,在母體毒性劑量400mg/kg/日(為人體暴露量的1100倍)時發現子代腎盂積水發生率增加。
哺乳大鼠給予佐米曲普坦後1小時,乳汁中藥物水平與母體血漿水平相當,4小時為血漿水平的4倍。
致癌性:
在大、小鼠上進行了佐米曲普坦灌胃給藥劑量達400mg/kg/d的致癌性研究。雌、雄小鼠給藥期限分別為92周和85周,最高劑量下的暴露量(原型藥物血漿AUC)約為人單次服用10mg(最大日推薦劑量)時的800倍,結果佐米曲普坦對腫瘤發生率沒有影響。大鼠試驗中,對照、低劑量、中劑量組大鼠給藥104~105周,高劑量組由於過高死亡率,給藥101周(雄性)和86周(雌性)後處死,結果僅在400mg/kg/日(約為人服用10mg後暴露量的3000倍)組出現雄性大鼠甲狀腺濾泡細胞增生和甲狀腺濾泡細胞腺瘤發生率增加。
藥代動力學
佐米曲普坦口服後吸收迅速,1小時內可達血藥濃度峰值的75%,隨後血漿濃度維持4~6小時。母體化合物的平均絕對生物利用度大約為40%。有一種活性代謝產物N-去甲基代謝物,也是一種5HTID激動劑。動物試驗結果表明,其效能為佐米曲普坦的2-6倍。健康成人給予單劑量佐米曲普坦後,在2.5—50mg劑量範圍內,佐米曲普坦及其活性代謝物的曲線下面積及血藥濃度峰值均與劑量成正比例。佐米曲普坦吸收不受食物的影響。
佐米曲普坦主要經肝臟生物轉換,然後代謝物從尿中排泄。三種主要代謝產物為:吲哚乙酸(血漿及尿中主要的代謝物)、N-氧化物及N-去甲基代謝物。N-去甲基代謝物有活性,其它代謝物無活性。N-去甲基代謝物的血漿濃度大致為母體藥物的一半;因此,預計其有助於本藥的治療作用。口服單次劑量的60%以上由尿中排泄(主要為吲哚乙酸代謝物),另約30%以母體化合物原形由糞便排泄。本品腎臟清除率大於腎小球濾過率,提示存在腎小管的分泌。
本品血漿蛋白結合率低(大約為25%),平均清除半衰期為2.5—3hr,其代謝物的半衰期也類似,提示它們的清除受轉換速率的限制。中度至重度腎臟損害的患者與健康受試者相比較,儘管母體化合物及其活性代謝物的曲線下面積僅有輕度的增高(分別為16%及35%),半衰期增長,但佐米曲普坦及其代謝物的腎臟清除率卻降低了(7—8倍)。佐米曲普坦多次給藥後不產生蓄積。
貯藏
密封、在乾燥處保存。
包裝
鋁塑包裝,2片/板/盒;2片/板×2盒。
有效期
24個月。
執行標準
YBH04932007
