
化合物簡介
基本信息
中文名稱:雷美替胺
中文別名:(S)-N-[2-(1,6,7,8-四氫-2H-茚並[5,4-b]呋喃-8-基)乙基]丙醯胺;雷美爾通;瑞美替昂;拉米替隆
英文名稱:Ramelteon;
英文別名:UNII-901AS54I69;Rozerem;Rozerem (TN);N-[2-[(8S)-2,6,7,8-tetrahydro-1H-cyclopenta[e][1]benzofuran-8-yl]ethyl]propanamide;
CAS號:196597-26-9
分子式:CHNO
分子量:259.34300
精確質量:259.15700
PSA:38.33000
LogP:2.95850
物化性質
外觀與性狀:結晶固體
密度:1.119g/cm3
熔點:113-115ºC
沸點:455.3ºC at 760 mmHg
閃點:229.2ºC
折射率:1.555
蒸汽壓:1.77EmmHg at 25°C
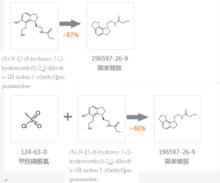
合成路線
 雷美替胺
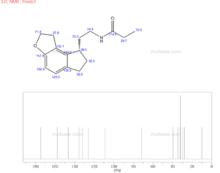
雷美替胺圖譜
1) 碳譜
 雷美替胺
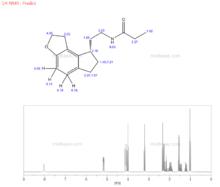
雷美替胺2)氫譜
 雷美替胺
雷美替胺藥效學和作用機制
本品為褪黑激素受體激動劑,與褪黑激素MT1和MT2受體有較高的親和力,對MT1和MT2受體呈特異性完全激動作用,而不與MT3受體作用。此外,它不與GABA受體複合物等神經遞質受體結合,在一定的範圍內也不干擾多數酶的活性,因此,能避免與GABA藥物相關的注意力分散(可能導致車禍、跌倒骨折等)以及藥物成癮和依賴性。其主要代謝物M一Ⅱ的總量是母體的20一100倍,但活性較低,與MT1和MT2受體的親和力分別約為母體的1/5和1/10。與原形藥物相比,其藥理活性降低約17—25倍。本品其他代謝物無活性。
藥動學
本品口服後顯示較強的首過效應,血清峰濃度(Cmax)和藥時曲線下面積(AUC)個體差異較大。空腹給藥吸收迅速,達峰濃度的中位值約為0.75(0.5~1.5)h,血漿蛋白結合率70%-82%,對紅細胞無選擇性分布。靜脈給藥後平均表觀分布容積約為73.6L。代謝時首先被氧化成羥基或羰基的衍生物,進而轉變成葡萄糖苷酸。本品在肝臟主要通過CYPlA2代謝,CYP2C亞族和CYP3A4也參與其代謝。本品呈單相快速消除,從尿液中可檢出其總量的84%,從糞便中可檢出4%,以原形排出體外的藥量不到0.1%。服藥後96h排泄基本完成。由於本品半衰期很短(平均約1—2.6h),qd多劑量給藥不會導致體內蓄積。與高脂餐同服時,AUC比空腹給藥高31%,Cmax降低22%,Cmax中位值約推遲45min,故應避免與高脂餐同服。
24例63—79歲的受試者單劑量口服本品16mg,平均Cmax為11.6ng/mL,AUC為18.7ng·h/mL,半衰期約為2.6h。AUC和Cmax分別提高97%和86%,這主要與老年人肝藥酶的活性減弱等有關。
本品對性別、輕中度肝腎功能不全以及慢性阻塞性肺病患者等特殊人群的主要藥動學和藥效學參數均無明顯改變,無症狀加重表現。嚴重肝功不全患者因代謝障礙,Cmax,AUC和半衰期明顯高於對照組,應禁用。
適應證和臨床評價
1.慢性失眠2項隨機雙盲試驗考察本品對慢性失眠的作用。年齡18-64歲,患有慢性失眠的患者分為平行服用單劑量本品或安慰劑兩組,持續35d,測定多導睡眠圖(PSG)。結果與安慰劑組相比,本品各測定時間點的睡眠潛伏期都有所縮短。另一項3周期的交叉試驗,年齡大於65歲並有慢性失眠史的患者,口服本品或安慰劑,結果與安慰劑相比,本品測定時間點的睡眠潛伏期也都有所縮短。說明對慢性失眠的療效肯定。
2.短暫失眠症一項隨機雙盲平行對照試驗考察了該藥物對短暫失眠症的首夜效應,健康成人受試者服用本品後測定其PSG。結果表明,與安慰劑相比,本品8mg即可縮短睡眠潛伏期。表明其對短期失眠療效確切。
安全性評價
1.藥物濫用14名受試者分別單劑量口服本品、三唑侖片和安慰劑,結果既使本品劑量達到推薦劑量20倍以上時,受試者藥物濫用傾向與安慰劑相比仍無差異,而陽性對照藥三唑侖則表現出穩定的量效關係。
2.藥物後遺效應:一項針對成年慢性失眠症患者為期35晚的雙盲、安慰劑對照試驗研究,在3個時間點測定了後遺效應。結果表明本品幾乎沒有次日後遺效應。
3.失眠症反彈/停藥後反彈在為期35d,針對苯二氮卓類受體阻滯劑停藥後20種常見症狀的自我問卷調查(BWSQ)顯示,服用本品的受試者BWSQ分值與安慰劑組類似,無反彈症狀。
4.內分泌功能:99名健康受試者口服本品16mg,qd或安慰劑,持續4周,結果未觀測到本品對內分泌功能有臨床意義的影響。在另一項臨床試驗中,122例失眠症患者服用本品16mg,qd或安慰劑,結果甲狀腺和腎上腺系統未見異常,但生殖系統出現異常反應,有19%出現泌乳激素水平增加,上升到4.9μg/L。一項開放研究顯示,2例出現清晨皮質激素水平異常以及後來的促腎上腺皮質激素刺激試驗反常。1例29歲女性患者被診斷為泌乳激素水平異常,但這些不良反應與本品的相關性尚不清楚。
不良反應
1.常見不良反應
1. 包括頭暈、頭痛、嗜睡、疲勞、失眠加重、抑鬱、關節痛、肌肉痛、胃腸道反應、味覺改變、上呼吸道感染、過敏反應等,且發生率和程度均較低,與安慰劑組相似,無嚴重不良反應。
現已明確,本品對成年人生殖系統激素水平有影響,如降低睪丸素水平和提高催乳素水平,但對青少年人群生殖系統的影響尚不清楚。若出現無法解釋的月經不調、乳漏,性慾下降或生殖問題,應考慮測定睪丸素水平和催乳素水平。
2.三致反應在SD大鼠中進行的致癌研究表明,本品劑量>250mg/kg/d時,雄鼠肝腺瘤和良性Leydig細胞瘤的發生率與劑量有關。劑量>60mg/kg/時,雌鼠肝腺瘤的發生隨劑量增加,良性Leydig細胞瘤的發生率也隨之增大,劑量在1000mg/kg/d時雄雌性鼠均會發生肝癌。嚙齒類動物使用非基因毒性藥物引起的Leydig細胞瘤,與睪丸素水平降低並伴黃體激素釋放的補償性增加相關,其中黃體激素刺激Leydig細胞增生。大鼠研究表明,日給藥劑量250-1000mg/kg/d,持續4周,將導致血漿黃體激素水平升高。但齧齒動物發生腫瘤的用藥濃度遠遠高於本品在人類最大推薦劑量(MRHD)時的平均血漿濃度,且大鼠Leydig細胞對黃體激素的反應比人體Leydig細胞更敏感。齧齒動物肝瘤和Leydig細胞瘤與人體腫瘤的相關性尚不清楚。
艾姆斯實驗、在鼠淋巴瘤TK+/-細胞系進行的體外哺乳動物細胞基因突變實驗、大鼠肝細胞體內/體外未排序DNA合成實驗以及大小鼠體內微核實驗均表明:本品無基因毒性。在大鼠肺細胞S9代謝活性存在情況下,本品在染色體畸變實驗中呈現陽性。
在再產卵和胚胎髮育早期給予SD大鼠口服本品,劑量達到600mg/kg/d對雌鼠和雄鼠的交配和排卵無影響。當雌鼠的用藥劑量≥60mg·kg~·d叫時,出現不規律的發情期,生殖細胞數降低,活胚胎數量減少。當劑量達到600mg/kg/d。時,黃體數量下降。持續7周給雄鼠劑量600mg/kg/d。時,對雄鼠的精子質量無影響,對於受精卵或胚胎也無影響。鑒於以上研究,雌鼠劑量為20mg/kg/d,雄鼠劑量為600mg/kg/d時,對於生殖終點沒有影響。研究顯示:本品劑量高於人體最高推薦劑量的197倍(以mg/m2為單位)才會顯示出一定的致畸作用。但尚未獲得本品在懷孕婦女中的研究數據,故應慎用。
藥物相互作用
1.肝藥酶激動劑/抑制劑對本品代謝的影響強CYPlA2酶抑制劑氟伏沙明100mgbid,連續服用3d,隨後單劑量服用本品16mg,可使本品的Cmax和AUC。分別增加約70倍和190倍。服用強CYP3A4酶抑制劑酮康唑200mg,bid,d4予本品16mg,結果本品的AUC。和Cmax與單用藥相比,分別增加了84%和36%。強CYP2C9酶抑制劑氟康唑能使單劑量服用本品16mg的AUC。和Cmax分別增加約150%。強CYP酶誘導劑利福平600mg,qd,持續11d,隨後單劑量服用本品32mg,結果本品及其代謝物M-Ⅱ的AUC。和Cmax平均降低80%。因此,本品與其代謝酶的激動劑/抑制劑聯用時應謹慎,需相應調整劑量,而強CYPlA2酶抑制劑如氟伏沙明則應禁止聯用。
2.本品對於其他藥物代謝的影響本品與奧美拉唑、右美沙芬、咪達唑侖、茶鹼、地高辛和滅鼠靈等合用時,無競爭抑制作用。
綜上所述,本品在治療失眠、入睡困難等方面療效顯著,且使用安全,治療窗寬,不良反應少,長期用藥不產生藥物依賴。其對人體的長期毒性、生殖毒性、致癌作用以及對青少年生殖系統分泌影響的研究也將不斷深入。
