疾病簡介
脊柱的冠狀位,矢狀位或軸向位偏離正常位置,發生形態上異常的表現,稱為脊柱畸形。脊柱側凸畸形特指在冠狀位偏離。分類
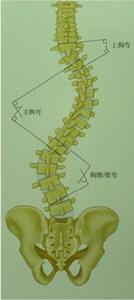
脊柱畸形根據位置可以分為頸椎,胸椎和腰椎畸形。根據形態學可以分為前凸,側凸和後凸畸形。根據脊柱畸形的原因考慮,可以分為特發性,先天性,神經肌肉型,間質性,創傷性等原因。對於側凸來說,特發性是其常見原因冠狀位畸形,將其畸形位置分為上胸段,中胸段,胸腰段/腰段。(圖1-1)
臨床表現
從外形上,側彎可以產生背部隆起畸形,產生“剃刀背”畸形,有的甚至產生“漏斗胸”或“雞胸”畸形,同時合併這種背部畸形,可以伴隨雙側肩關節不平衡或者骨盆不平衡,以及雙下肢不等長,可以引起患者明顯局部畸形,身高減少,胸腔和腹腔容量的減少,甚至造成神經功能,呼吸功能,消化功能的損害等;同時對於脊柱骨結構本身發育不良的患者,可以伴發腦脊膜膨出,隱形脊柱裂等神經發育異常的表現。此外,先天性脊柱側凸還可能伴有心血管系統異常,氣管-食管瘺,多囊腎等多臟器異常的表現影像學測量
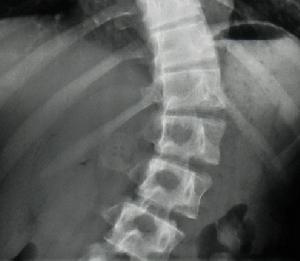
影像學方面對於脊柱側凸或者後凸的診斷標準,包括Cobb角的測量,即選擇組成側凸或者後凸兩端(最頭端和最尾端)最傾斜的椎體之間成角,是對於任何脊柱畸形最基本的描述。(圖1-2)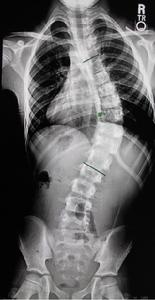
對於椎體鏇轉發生的評價,通常用Nash-Moe分型,即通過雙側椎弓根出現的對稱性多少來判斷。通過椎體椎弓根雙側的對稱性描述,從而在普通X線上得到椎體鏇轉的信息;(圖1-3)
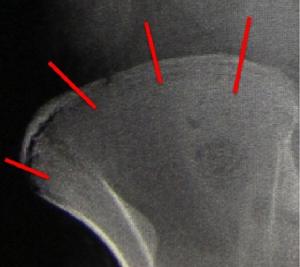
以及對於患者生長潛力的評價Risser征等。Risser征是通過雙側髂骨表面骨骺閉合的情況,評價患者的生長潛能。(圖1-4)
特發性脊柱側凸
病因
特發性脊柱側凸,從病因學上來講,並不十分明確,但是和基因和遺傳具有一定關係,此外還存在椎旁肌肉本身分布不平衡的原因。形態學是指椎體本身沒有結構異常,椎體分隔正常,擁有對稱的椎弓根,發育正常的椎板和關節突。分類
從發病時間上進行分類,可以將其分為嬰幼兒,少年,青少年和成年四種。嬰幼兒是指發病在0-3歲,少年是指發病年齡在4-10歲,通常是在青春期前;青少年發病年齡在10歲-骨骺閉合的青春期,是成年前脊柱側凸最常見類型,;而成人特發性脊柱側凸,是指青少年期間形成的脊柱側凸,由於沒有進行治療,或者進行了一定治療,但是畸形沒有明顯改善,進入成年期有進一步進展的側凸。臨床表現
特發性脊柱側凸最常見“剃刀背”(圖2-1),某些患者還會發現雙側肩關節不平衡和骨盆不平衡。通常很少會發生神經損害的表現。值得注意的是某些神經纖維瘤病,或者先天性脊柱側凸,會出現類似“特發性脊柱側凸”的曲度,手術前必須對於脊髓功能進行明確評價。治療
分為手術治療和保守治療兩種方法:特發性脊柱側凸進行手術的標準存在一定爭議,多數學者認為側彎Cobb角度在45°以上,或者出現肩關節或者骨盆失平衡,可以考慮手術治療,手術方式可以分為側前方手術和後路手術進行固定矯形植骨融合。
保守治療可以分為觀察和支具治療,對於可以觀察的患者,通常認為具備一定生長能力(即Risser征小於3),同時局部Cobb角度小於25°,或者Risser征為4或5,脊柱已經停止生長,但是局部Cobb角度沒有達到手術標準;
支具治療,是指脊柱具備一定生長能力(Risser征小於3),局部Cobb角度位於25°至45°之間的患者。對於支具的選擇,可以根據側凸頂椎的位置選擇相應類型,一般側凸頂椎位於T7水平以上,可以選擇Milwaukee支具,而位於T7水平以下可以選擇Boston支具。圖2-1:特發性脊柱側凸背部“剃刀背”
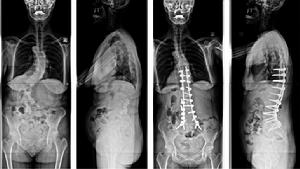
圖2-2:特發性脊柱側凸手術前後對比:男,22歲,術前主胸彎62°,術後矯正為23°
圖2-3:女性,14歲,特發性側凸,胸腰段角度為23°,主胸彎角度22°,支具固定1年複查,佩戴支具時角度為17°
先天性脊柱側凸
對於先天性脊柱側凸或者後凸,通常是指椎體本身結構異常而導致的脊柱畸形。其病理結構類型,通常分為椎體骨骼形成不全(Defect of formation)或者分隔不全(Defect of segmentation)。椎體形成不全的最大特點是其椎間隙通常正常存在,但是椎體一側椎弓根發育不全或者消失,或者和鄰近椎體發生融和,或者X線顯示椎體椎間隙距離增寬;椎體分隔不全最大的特點是兩椎體間(甚至多個椎體間)的椎間隙消失,椎體融合為一個整體的同時,一側椎弓根消失,甚至形成了骨性連線(bone bar),對側可以是半椎體形成伴有椎間盤發育。這種情況可以發生在冠狀位或者矢狀位,從而造成了脊柱側凸或者後凸。
先天性脊柱畸形,通常伴有脊髓的畸形,諸如椎管內腫瘤,脊髓縱裂(5-21%),椎管內骨(膜)性縱隔形成,脊髓栓系,低位圓錐,脊髓空洞,腦脊膜膨出等,同時椎管內也會發生表皮樣囊腫,皮樣囊腫,畸胎瘤等良性腫瘤。造成此種脊髓和脊柱畸形伴隨出現的原因,是由於在胚胎髮育過程中(胚胎髮育前6周)形成的脊索和髓節發育異常。在臨床表現上可以表現腰骶部竇道,汗毛增多,局部隆起,皮下脂肪瘤等。
先天性脊柱畸形通常伴有其他臟器的發育畸形,可以用VACTERL來縮寫,即為V-椎體發育畸形,A-肛門閉鎖,C-心血管畸形,TE-氣管食管瘺,R-腎臟發育不良,L-肢體發育不良。同時還可能伴發翼狀肩胛(Sprengel Deformity),Klippel-Feil綜合徵等病變。
對於先天性脊柱側(後)凸的治療,分為觀察和手術治療。支具治療對於這類患者是沒有作用的,因為支具矯正力是無法糾正椎體存在的先天性畸形。如果患者無明顯外觀異常,諸如肩關節和雙髖不對稱,背部的畸形,自己沒有不適感覺,可以考慮繼續觀察,時間大約是4-6個月;反之可以考慮手術治療,手術根據不同年齡,畸形位置和全身平衡情況,可以選擇不同的手術方式。諸如半椎體骨骺融和術,半椎體切除,側凸凸側原位融合術等。目前隨著手術器械和理念的改進,對於先天性半椎體的治療,主要考慮對半椎體進行切除,內固定矯形融和手術。先天性脊柱側彎在任何年齡均會造成畸形的進展。
圖3-1:椎體分隔不全和成型不全的圖示說明
圖3-2:腰3左側先天性半椎體,行手術切除後
圖3-3:椎管內骨性縱隔
圖3-4:椎管內二分脊髓
神經肌肉型脊柱側凸
定義和分類神經肌肉型脊柱側凸,主要是由於全身肌肉系統病變,導致胸背部本身肌肉無力和病變所造成椎旁肌不能夠很好的支撐脊柱所造成。脊柱側凸研究協會(Scoliosis Research Society, SRS)將其分為上運動神經元損害(諸如腦癱,脊髓空洞症,脊髓損傷),和下運動神經元損害(諸如小兒麻痹症,脊柱肌肉萎縮等)。臨床表現其中最常見的原因就是腦癱。腦癱多數是由於新生兒時期缺氧所造成神經系統發育不良,導致肌肉持續性萎縮和發育不平衡。同時,此類患兒區別於其他類型側凸,經常伴隨全身疾病,例如關節脫位,癲癇,智利障礙,甚至是褥瘡。發病時間多數是在嬰兒期或少年期就開始,脊柱骨骼本身發育良好,椎體外形無變異,但是由於雙下肢不能夠行走,多數患兒只能做在輪椅上,伴有明顯的雙下肢肌肉萎縮,髖關節容易出現一側內收攣縮,和另一側外展脫位同時存在。同時,這種肌肉萎縮可以進一步影響支撐呼吸功能的肋間肌(例如Duchenne綜合徵),引起早期死亡。
脊柱畸形特點脊柱外形通常呈現明顯的“C“型曲度,自頸椎-胸椎-腰椎連續變化,沒有明顯的代償側凸形成。側彎曲度大,影響節段常,脊柱柔軟性差,常伴有明顯的骨盆傾斜。
治療原則全身情況,骨科專科情況和脊柱畸形同時進行評價。對於此類病人,具有支撐功能的座椅十分必要,支具治療在青春期生長高峰來臨後基本無效,手術的固定通常要從上胸椎(T1或者T2)融合到骨盆。
圖4-1:小兒麻痹症引起脊柱側凸 ,腦癱引起脊柱畸形,unirod固定矯形術後
神經纖維瘤病
神經纖維瘤病也是造成脊柱側(後)凸的重要原因。神經纖維瘤病本身是由於基因缺陷導致神經嵴細胞發育異常導致多系統損害。根據臨床表現和基因定位分為神經纖維瘤病I型(NFI)和II型(NFII)。NFI患者查體時通常皮膚表面可以看到牛奶-咖啡斑(Café au lait)和周圍神經多發性神經纖維瘤,多位於軀幹非暴露部位。另外眼部可見Lisch結節,是上瞼纖維瘤或者叢狀神經纖維瘤,眼眶可觸及腫塊或者凸眼搏動,裂隙燈可見虹膜粟粒橙黃色圓形小結節,為錯構瘤,是NFI特有表現,可隨著年齡而增大。診斷標準是青春期前6個以上,直徑大於5mm(青春期後直徑大於15mm)具有高度診斷價值;全身和腋窩雀斑也是特徵之一;發現2個或者2個以上神經纖維瘤或叢狀神經纖維瘤;以及 親屬中有NFI患者;2個或2個以上Lisch結節;骨損害。基因病變位置多位於17q11.2。
NFII患者多為中樞神經纖維瘤或者雙側聽神經瘤病,診斷標準一級親屬患有NFII伴一側聽神經瘤,或伴有神經纖維瘤,腦脊膜瘤,膠質瘤,Schwann細胞瘤中的兩種。基因病變位置位於22q。
神經纖維瘤病I型容易導致脊柱側(後)凸畸形,患者發病早(通常在青春期前),側(後)凸進展快,畸形明顯,曲度僵硬,骨質本身強度降低等。支具治療對於畸形的糾正幾乎沒有作用。
對於神經纖維瘤病I型的治療原則,應該將患者分為無發育營養不良和發育營養不良兩組。前者的脊柱曲度,類似特發性側凸的外形,處理原則,諸如融和節段也相似。但是實際的角度和骨質方面,兩者則明顯不同,神經纖維瘤病曲度進展明顯,同時術後假關節形成多,椎體發育方面可見缺陷。對於小於35°無發育營養不良組,可行試驗性支具治療;35°到45°可行單純後路手術治療;60°以上可行前後路聯合手術,以增加融合率。發育營養不良組通常具有椎體“貝殼”樣變,椎體嚴重鏇轉,椎弓根距離增加,凸側肋骨呈現“鉛筆”征等表現,和椎管內腫瘤和硬膜增寬有關。曲度的進展多在7歲之前發病。支具治療完全失效。由於曲度大,椎體發育不良,通常手術治療不能夠提供足夠力量控制畸形進展,並且容易形成局部假關節和術後曲度進展。對於營養不良和非營養不良造成側後凸,假關節發生率無顯著區別,但是進行360°融合可以提高融合率,尤其是形成側後凸的患者,避免畸形的進展。和其他類型側凸相比,神經纖維瘤病更加傾向早期矯正融和,而對於軀幹生長影響不大。
神經纖維瘤病I型也非常容易引起頸椎後凸畸形,引起局部疼痛,同時容易被忽略。
圖5-1:神經纖維瘤病引起胸腰段側凸
圖5-2:手術矯形固定術後
Marfan(馬凡)綜合徵
Marfan綜合徵也是引起脊柱側凸的原因,男女發病比例類似,是染色體顯性遺傳病(15號染色體15q21.1變異造成),但是也有約25%的患者是染色體變異造成的。Marfan綜合徵也稱為蜘蛛掌畸形。其脊柱畸形發病年齡早,50%的患者是6歲時首發的,儘管外觀類似特發性側凸表現,但是Cobb角大,進展快,曲度僵硬,不易進行矯正,同時容易出現椎體間的側方位移,植骨容易出現不癒合。診斷標準
現代的診斷標準是在1986年第七屆人類遺傳學國際大會上建立,並在1988年第一次國際馬凡綜合徵專題討論會上明確規定的標準,1996年,de Paepe A重新修正被視為統一標準。1.標準的具體內容
⑴骨骼系統
主要標準:以下表現至少有4項——雞胸;漏斗胸需外科矯治;上部量/下部量的比例減少,或上肢跨長/身高的比值大於1.05;腕征、指征陽性;脊柱側彎大於20度,或脊柱前移(側彎計);肘關節外展減小(<170度);中踝中部關節脫位形成平足;任何程度的,髖臼前凸(髂關節內陷)(X片上確定)。
次要標準:中等程度的漏斗胸:關節活動異常增強;高齶弓,牙齒擁擠重疊;面部表征:長頭——正常頭顱指數為75.9或以下、顴骨發育不全、眼球內陷、縮頜、瞼裂下斜。
骨骼系統受累需符合的條件:至少有兩項主要標準或一項主要標準加兩項次要標準。
⑵眼睛系統
主要標準:晶狀體脫位。
次要標準:異常扁平角膜(角膜曲面計測量);眼球軸長增加(超聲測量);虹膜或睫狀肌發育不全致瞳孔縮小。眼睛系統受累需符合標準:主要標準或至少兩項次要標準。
⑶心血管系統
主要標準:升主動脈擴張伴或不伴主動脈瓣返流,以及至少Valsava氏竇擴張;升主動脈夾層。
次要標準:二尖瓣脫垂伴或不伴二尖瓣返流;主肺動脈擴張(在無瓣膜或外周肺動脈狹窄及其它明顯原因下,年齡又小於40歲);二尖瓣環鈣化(年齡小於40歲);降主動脈或腹主動脈擴張或夾層(50歲以下)。
心血管受累需符合的條件:有一項主要標準或一項次要標準即可。
⑷肺系統
主要標準:無。
次要標準:自發性氣胸;肺尖肺大泡(胸片證實)。
如果一項存在即可認為肺系統受累。
⑸皮膚和體包膜
主要標準:無。
次要標準:皮紋萎縮(牽拉痕),與明顯超重、妊娠或反覆受壓等無關;復發性疝或切口疝。
一項次要標準存在即可認為皮膚或體包膜受累。
⑹硬腦(脊)膜
主要標準:CT或MRI發現硬脊膜膨出。
次要標準:無。
⑺家族或遺傳史
主要標準:父母、子女或兄弟姊妹之一符合該診斷標準;FBNI基因中存在已知的導致馬凡綜合徵的突變;存在已知的與其家族中馬凡綜合徵患者相同的FBNI基因單倍型。
次要標準:無。
由於家族或遺傳史在診斷中意義重大,主要標準中必須有一項存在。
2.診斷前提:Marfan綜合徵和Marfan體型是在診斷時容易混淆的
Marfan綜合徵的診斷:
對特定病例:如果無家族或遺傳史者,至少需有兩個不同系統的主要標準以及另外一個器官受累;如果檢出一個已知Marfan綜合徵的基因突變,一個系統中有一項主要標準和第二項系統受累即可診斷;具有家族史患者,具有一個系統的一項主要標準和第二個系統受累即可診斷。
Marfan體型疾病的診斷
具有類似臨床表現,但是沒有符合以上診斷標準。Marfan綜合徵 | Marfan體型 | |
發病過程 | 脊柱畸形發病率52%-100%。容易造成呼吸功能衰竭。曲度進展可以超過180° | 脊柱畸形發病率34-88% |
表現 | 位於曲度部位的疼痛 | 多無疼痛 |
曲度類型 | 多為雙結構型側凸,單彎多為右側胸彎 | 多為單彎(以右胸彎和胸腰彎為主) |
曲度角度和進展 | 進展快 | Marfan綜合徵類似 |
嬰幼兒脊柱側凸 | 發病早,多為嬰幼兒和少年期,無法自行糾正 | 發病年齡多為8.7-10.5歲 |
治療
支具治療對於Marfan綜合徵或Marfan體型疾病無效。手術治療主要是通過後路固定矯形融合的方法,必要時聯合前路手術椎間隙植骨融合。術前評估是非常重要的,尤其是Marfan綜合徵容易並發心肺主動脈等重要臟器的病變,因此對於術前的整體評估不可缺少。
圖6-1:蜘蛛掌
圖6-2:膝關節活動度增大
圖6-3:關節韌帶鬆弛導致活動度增加
圖6-4:關節韌帶鬆弛導致活動度增加
圖6-5:全脊柱正位X線和CT
成人脊柱側凸
成人脊柱側凸主要存在兩種病理類型,其一是由於青少年時期的特發性脊柱側凸進展到成人期而出現相應症狀,稱成人特發性脊柱側凸;其二是在成年期內由於椎間盤退變造成,稱退變性成人脊柱側凸(De novo Scoliosis)。後者是最常見的類型。另外還包括先天性脊柱側凸成年表現,麻痹性側凸,外傷後畸形等。成人脊柱側凸的發生率可以達到2.9%。
臨床表現以疼痛為主要表現形式,同時會伴有腰椎管狹窄的症狀。對於病史的詢問,應該包括對於日常功能的評價,脊柱畸形對於工作,生活的影響。查體包括對於脊柱畸形,肌肉骨骼系統和神經系統的檢查,包括雙下肢的長度,以避免矯形後重新造成身體的不平衡。
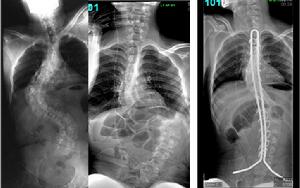
影像學檢查包括全脊柱正側位,動力位(左右Bending像)來明確間盤活動性,通常成年特發性側凸和成人退變性側凸在影像學上的區別在於:1,前者通常有兩個曲度,後者通常只有一個;2,前者軀幹和骨盆代償良好,後者常見失代償現象;3,前者椎間隙無明顯退變,椎間隙等高且無明顯矢狀或冠狀位滑移;後者椎間隙退變嚴重,雙側不等高,可見明顯滑移,以及終板增生的改變等;CT明確骨質變化情況,尤其是伴有嚴重骨質疏鬆患者;MRI用來明確間盤退變情況和神經壓迫情況,有的學者甚至使用間盤造影來明確遠端融和間盤的水平。
鑒於其患者對象的特殊性,其手術併發症的發生率可以達到40%。術前需要對於患者整體情況進行評估(包括心腦血管功能,肺功能等)。
非手術治療,主要是進行功能鍛鍊,使用N-saids消炎止痛藥物緩解症狀,使用支具給予一定的支撐作用,非手術治療不能夠緩解畸形的進展。
手術治療的指征:1,畸形進展;2,脊柱平衡功能差;3,畸形嚴重影響心肺代償功能;4,具有神經功能的損害。
圖7-1和7-2退變性脊柱側凸術前正側位;圖7-3和圖7-4術後固定情況