
疾病分類
 神經纖維瘤病
神經纖維瘤病發病原因
神經纖維瘤病是常染色體顯性遺傳病。NF1其致病基因位於常染色體17q11.2。在發病者,此染色體位點缺失,致使患病者不能產生相應的蛋白-神經纖維瘤蛋白。神經纖維瘤蛋白是一種腫瘤抑制因子,通過加快降低原癌基因p21-ras(在細胞內有絲分裂信號轉導系統中起主要作用)的活性從而減緩細胞增殖。NF2致病基因定位於常染色體22q11.2。患病者此基因位點缺失,致使患者體內不能產生雪旺氏細胞瘤蛋白。該蛋白是否是抑癌基因及其作用機制目前尚不清楚。但它可能在細胞周期的運行、細胞內及細胞外信號轉到系統中起作用。
發病機制
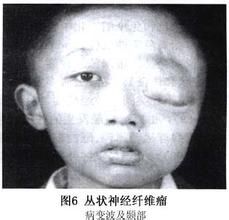
神經纖維瘤由雪旺氏細胞和成纖維細胞組成。其細胞外機制嵌入神經束膜細胞、軸突和肥大細胞。叢狀神經纖維瘤病和皮膚神經纖維瘤病的細胞組成相同。但是叢狀神經纖維瘤病有更為廣泛的細胞外基質,而且往往含有豐富的血管網。神經纖維瘤病和累計多個神經或神經束,向周圍結構延伸從而導致相應的功能障礙以及軟組織和骨結構的增生。叢狀神經纖維瘤病偶爾會惡變成紡錘細胞瘤(周圍神經鞘惡性腫瘤)。每種細胞類型在神經纖維瘤病的發生、發展過程中扮演的角色仍不清楚。
病理生理
神經纖維瘤病主要的致病機制為腫瘤生長對周圍組織破壞產生症狀,如消化道出血等;腫瘤增長本身對相應的周圍神經產生壓迫出現相應的神經功能障礙如麻木,肌無力等;腫瘤生長於顱內,產生占位效應導致顱內壓增高產生頭痛、嘔吐等症狀;或腫瘤刺激腦組織產生異常放電形成癲癇等。
臨床表現
神經纖維瘤病1型的臨床表現
1)牛奶咖啡斑:幾乎所有的患者都有皮膚色素斑,呈淡棕色、暗褐色或咖啡色。腋窩部出現雀斑樣色素沉著,生理變化如發育、妊娠、絕經、精神刺激均可使之加重,有時皮疹出現較遲,在發育期才開始發病,緩慢發展。
2)多發性神經纖維瘤:患者常訴全身出現無痛性皮下腫物,並逐漸增加和擴大。青春期和妊娠期明顯進展。多無臨床症狀。少數表現為放射性或灼燒樣疼痛,腫瘤壓迫視神經引起視力下降等。
3)神經症狀:多數患者無不適主訴,僅少數患者出現智力下降、記憶力障礙、癲癇發作、肢體無力、麻木等。
4)骨骼損害:少數患者出生時即出現骨骼發育異常,或腫瘤生長過程中壓迫骨骼引起異常。
5)內臟損害:生長於胸腔、縱膈、腹腔或盆腔的神經纖維瘤可引起內臟症狀,其中消化道受累可引起胃腸出血或梗阻,還可引起內分泌異常。
神經纖維瘤病2型的臨床表現
首發症狀以雙側進行性聽力下降最為常見,亦有部分病人表現為單側嚴重的聽力障礙或波動性聽力喪失或突發性聽力喪失。最常見的臨床表現為耳鳴、聽力下降,頭暈,眩暈這少見,其次為手顫、走路搖擺、語調異常等共濟失調表現,口角歪斜,面部麻木感等,這些症狀多為單側。少數患者訴持續性頭痛,伴噁心、嘔吐和視物不清等顱內壓增高表現。
診斷
輔助檢查
1)超聲檢查:可見多發實質性腫塊,可位於皮下、腹腔、盆腔等。
2)眼科檢查:通過裂隙燈可見虹膜粟粒狀、棕黃色圓形小結節,也成為Lisch結節或虹膜錯構瘤。眼底鏡可能發現顱內壓增高導致的視乳頭水腫或視神經萎縮。
3)CT和MRI:對於脊柱內或顱內的腫瘤可通過CT或MRI檢查發現。腫瘤在CT密度通常較脊髓和腦組織略高,呈圓形或類圓形。在MRI上神經纖維瘤表現為T1上低或等信號,T2上高信號。部分腫瘤伴有囊變。增強掃描後腫瘤多明顯強化。
4)神經電生理檢查:表現為神經源性損害,電信號傳導減慢等。
診斷
1987年美國NIH制定了診斷標準:
NF1:
1)6個或以上的牛奶咖啡斑,青春期前最大直徑5mm以上,青春期後15mm以上;
2)2個或以上任意類型神經纖維瘤或1個叢狀神經纖維瘤;
3)腋窩或腹股溝褐色雀斑;
4)視神經膠質瘤;
5)2個或以上Lisch結節,即虹膜錯構瘤;
6)明顯的骨骼病變:如蝶骨發育不良,長管狀骨皮質菲薄,伴有假關節形成;
7)一級親屬中有確診NF1的患者。
上述標準符合2條或以上者可診斷NF1。
NF2
1)雙側聽神經瘤;
2)有NF2家族史(一級親屬中有NF2患者),患單側聽神經瘤;
3)有NF2家族史(一級親屬中有NF2患者),患者有以下病變中的2中:神經纖維瘤、腦膜瘤、膠質瘤、雪旺氏細胞瘤、青少年晶狀體後囊渾濁斑。
上述標準符合1項即可診斷NF2。
鑑別診斷
NF1需要和以下疾病鑑別:
1)結節性硬化也是常染色體先行遺產並的神經皮膚綜合徵,累計皮膚和神經系統。皮膚該表包括口鼻三角區對稱性蝶形分布的皮脂腺瘤、葉狀白斑、鯊魚皮斑,也可見牛奶咖啡斑;神經系統損害以顱內結節性鈣化灶為特徵性表現,臨床上常見難治性癲癇和智慧型減退;眼科檢查可見視乳頭附近蟲卵樣鈣化節節或視網膜周邊黃色環狀損害。
2)McCune-Abright綜合徵罕見的先天性疾病,以骨纖維發育異常為主,可見骨皮質變薄、容易發生病理性骨折,鹼性磷酸酶增高;伴皮膚大片的咖啡樣色素沉著,以及內分泌疾病,如甲亢、甲旁亢、性早熟、Cushing綜合徵等。一般不累及神經系統,智力正常。
3)Proteus綜合徵多種組織非對稱性過度生長造成的畸形。特徵性的表現是組織痣,呈黃褐色或黑褐色,邊界清楚,隆起呈鵝卵石質地,常有條紋狀外觀,位於足趾、手掌、手指、鼻部和軀幹,病例活檢示角化過度、棘層肥厚及乳頭瘤樣增生。神經系統損害主要表現為智力障礙,眼科表現包括眼球外層皮癢囊腫和眼球上囊腫。
NF2需要和以下疾病鑑別:
1)其他前庭和中樞神經系統疾病引起的眩暈表現為視物鏇轉,伴噁心、嘔吐、出汗等自主神經症狀。NF2的腫瘤包裹、壓迫前庭神經,由於腫瘤生長緩慢,病人多可逐漸代償,因此病人較少出現真性眩暈,可僅表現為頭暈和行走不穩感。NF2往往伴有雙側聽力進行性下降。
2)發生於橋腦小腦角區的腫瘤常見的為腦膜瘤、膽脂瘤及三叉神經鞘瘤等,腦膜瘤有典型的腦膜尾征,膽脂瘤和三叉神經鞘瘤可產生三叉神經痛等症狀。
3)轉移瘤腦內伴發的多發性病灶要與轉移瘤鑑別,後者大多有原發病變,結合臨床和病史可鑑別。
疾病治療
NF1的治療
1)一般不需要治療;
2)對症治療:如病人出現放射性或灼燒樣疼痛難以忍受時,可復用陣痛藥物;繼發症狀性癲癇的可給予藥物抗癲癇治療。藥物首選卡馬西平,起始劑量可0.2克,一日兩次,根據藥物濃度和治療效果逐漸調整藥物劑量,用藥期間注意監測病人血象和血生化指標;
3)手術治療:
a)小兒局限的神經纖維瘤可一次切除;
b)巨大的腫瘤可全部或部分切除。
c)叢狀神經纖維瘤病變部位多有豐富的血管網,術中應該注意在病變周圍正常組織處切口,徹底切除腫瘤及其周圍組織,術後可用雷射照射,防治復發。
d)單側眶板缺如可修補。
NF2的治療
手術治療是目前有效的首選的治療方法。手術多採用枕下乙狀竇後入路,在電生理檢測下仔細辨別面聽神經的位置,儘可能解剖和功能保留上述神經。術中往往需要磨除部分內聽道的後壁以期達到腫瘤全切除。
疾病預後
目前尚無有效的措施能阻止或逆轉NF1的病程。但是NF1疾病本身多僅影響面容,不影響正常壽命,除非良性腫瘤影響了重要臟器的功能。
NF2的預後較差,雙側手術切除後往往導致雙耳全聾,而次全切除術後復發率高。轉歸主要有死亡、聽力喪失及面癱。但是在現代顯微神經外科的理念以及術中電生理監測的幫助下,術中解剖或功能保留面聽神經、全切腫瘤的機會也越來越大。
疾病預防
本病尚無有效的預防措施。
疾病護理
神經纖維瘤病很少惡變,病人可保持良好的心理狀態。合理飲食,加強營養,適當運動,有助於增強免疫力。有神經功能障礙者建議在康復醫師的指導下進行有序的康復訓練。定期隨訪,一般應3-6個月隨訪複查一次,可根據具體情況延長或縮短隨訪時間。
