
發病部位
郎格罕細胞組織細胞增生症(Langerhanscellhistiocytosis,LCH)曾稱為組織細胞增生症X
(HistiocytosisX,HX),是一組由郎格罕細胞(Langerhanscell,LC)為主的組織細胞在單核-巨噬細系統廣泛增生浸潤為基本病理特徵的疾病。本病好發於骨、肺、肝、脾、骨髓、淋巴結和皮膚等部位。傳統上本病分3型:勒雪氏病(Letterer-Siwedisease,LS)、韓-雪-柯病(Hand-Schüller-Christiandisease,HSC)、骨嗜酸細胞肉芽腫(Eosinophilicgranulomaofbone,EGB)。
病因
尚未明確,多數學者認為本病為分化性組織細胞增生症,屬介於免疫反應性非腫瘤性增生和惡性腫瘤性組織細胞增生疾患之間,也有認為是一種惡性克隆性疾病。多數研究表明,LCH的發生可能與免疫缺陷有關,CD4減低、CD4/CD8比值下降,臨床分型(分級)>Ⅲ級者,惡性程度較高,故本病介於良性與惡性腫瘤之間。近有研究者,用流式細胞儀(Flowcytometry.FCM)分析病變區細胞DNA含量,發現有惡性腫瘤標誌的非正倍體,且S期細胞比例增加,推測是郎格罕細胞的惡性增殖所致。此外,本病可與惡性淋巴瘤同時存在,故認為部分LCH具有惡性性質。
有研究表明可能人類白細胞抗原與本病有一定的關係,國內有報告發現LCH患兒HLA-DR3和HLA-B40的抗原(基因)呈顯著增高,提示它們可能為易感基因,與LCH的發病有一定關係。
病變部位由於浸潤的病理組織細胞(郎格罕細胞)過度增殖,並產生白細胞介素-1(IL-1)和前列腺素E2(PGE2),從而引起臟器和組織的損害。骨病變部位的郎格罕細胞,通過局部分泌產生過量的IL-1和PGE2,可活化與促進破骨細胞的功能而引起骨質吸收,產生溶骨性損害。
臨床表現
根據其類型不同,其臨床症狀和體徵表現多樣。
發熱
熱型不規則,可為持續性高熱或間斷高熱。一般用抗生素無效。
皮疹
皮膚可因郎格罕細胞浸潤出現特異性皮疹,皮疹為多形性,皮疹多在胸背部和頭皮、髮際和耳後,起初為針尖到粟粒大小紅色斑丘疹,以後類似於濕疹或脂溢性樣滲性皮疹,多為出血性,然後結痂、脫屑,殘留色素白斑。各期皮疹同時存在或成批出現。
肝脾淋巴結腫
肝脾可中度至重度腫大,脾大較肝大顯著。可見全身淋巴結輕度腫大,少數患嬰可伴有胸腺腫大。發熱、皮疹和肝脾腫大有伴隨關係。發熱、出疹時,肝脾增大;疹消、熱退,肝脾縮小。
骨質損害
X攝片的病變特徵是溶骨性骨質破壞,骨缺損系由於郎格罕細胞在骨質內增生浸潤所致。骨缺損局部如顱骨的頭皮呈包塊狀突起,可有輕壓痛,當病變侵蝕骨外板時,腫塊變軟而有波動,常可觸及顱骨缺損的邊緣。
1.顱骨缺損:最多見,顱骨呈地圖樣缺損,最早出現於頂、枕、眼眶、顳骨岩部、下頜骨等處。
2.下頜骨破壞:發生率(7%),頜骨病變可分為牙槽突型和頜骨體形兩種,前者首先侵犯
牙槽突,後向根尖下發展,破壞下頜骨體或上頜竇底,重者使牙齒完全失去支持骨呈“漂浮樣”改變,後者常開始於下頜骨體中央,漸波及牙槽突、下頜骨下緣或升支後緣。頜骨病變伴隨口腔頜面臨床改變有兩面種表現:①牙齦或齶部的潰瘍性病損,可伴有腫脹和周圍黏膜增生。②頜面腫脹或包塊,可伴有牙齒鬆動或疼痛,重者可造成牙齒脫落。
3.顳骨與乳突破壞:中耳道有肉芽腫病變引起顳骨與乳突破壞,常有耳流膿,乳突腫脹或凹陷。
4.眼球突出:由於眼眶骨受損引起,常為一側,也可雙側突出。5.尿崩症:由於蝶胺破壞與組織細胞浸潤累及垂體或下丘腦所致,表現為多飲、多尿。
6.其他長骨等破壞:組織細胞浸潤累及骨盆、肩胛、脊椎、肱骨、股骨等引起溶骨性破壞,局部表現腫脹或發生病理性骨折。
肺部浸潤
多見,年齡越小越容易受累,常有咳嗽、氣促,重者有發紺。極易發生肺炎或肺泡破裂,可形成大小不等的肺泡囊腫或出現氣胸、皮下氣腫,嚴重者可發生呼吸衰竭而死亡。
耳
表現為外耳道溢膿、耳後腫脹和傳導性耳聾。
骨髓
骨髓中可出現郎格罕細胞,侵犯骨髓者常有貧血、粒細胞減少和血小板減少。(八)中樞神經系統:最常受累的部位是丘腦-垂體後區,常見症狀有尿崩症,還可有共濟失調、構音障礙、眼球震顫等神經系統等症狀
檢查
實驗室檢查
(一)血象:無特異性改變,多器官受累者常有中度以上貧血,可有白細胞減下降和血小板減少。
(二)骨髓象:部分病例有骨髓增生低下,可見組織細胞增多,但罕見嗜血現象。有骨髓受累的患者常伴有貧血、白細胞減少,以及發熱、皮疹等表現。但骨髓中組織細胞數量與骨髓功能異常並無正比關係。
(三)尿比重測定:如尿比重常在1.001~1.005,或尿滲透壓<200mOsm/L,則提示可能有蝶胺破壞與組織細胞浸潤累及垂體或下丘腦所致。
(四)免疫功能檢測:⑴體液免疫:除IgM常增高外,大都正常。⑵細胞免疫:CD3多減低,CD4/CD8降低或增高,可有淋巴細胞轉化功能降低,T淋巴細胞組胺H2受體缺乏。
影像檢查
(一)X線檢查:
1.胸部攝片:胸部X線見肺部有網點狀陰影,或呈毛玻璃狀,或在此基礎上出現顆粒狀陰影,嚴重者出現囊狀陰影、肺氣腫和氣胸等。
2.骨骼攝片:顯示溶骨性骨質破壞,一般應多部位照片,依次為頭顱、脊柱、骨盆和四肢。(二)CT或MRI掃描:雙側顳骨CT或MRI掃描,以明確蝶骨各部分、蝶鞍骨質與垂體等受損害情況。(三)ECT全身骨骼系統掃描,可檢出骨損害的部位與大小。五、病理組織學檢查
病理組織檢查
病理檢查是確診郎格罕細胞組織上細胞增生症的主要手段。故應儘可能做活組織檢查,皮疹穿剌液印片和皮膚活檢最常用,有淋巴結腫大者可做淋巴活檢,骨質缺損做腫物刮除時做刮除物檢查。
(一)光鏡:病變部位(皮膚、淋巴結、骨髓等)見到特徵性的分化較好的郎格罕組織細胞(單核的組織細胞、泡沫細胞)增多可以確診,郎格罕細胞特徵為細胞核為單個或多個,核摺疊,有核仁。
(二)免疫組化:病變細胞的免疫組化CD1a單抗染色陽性為診斷的重要依據,免疫組織化學染色—S-100神經蛋白(Neuroprotein)陽性,α-D-甘露糖苷酶陽性,可與花生凝集素結合。
(三)電鏡:有條件時應作電鏡檢查,病變細胞內找到有Birbeck顆粒的郎格罕細胞。
診斷
LCH的診斷:此症傳統的診斷方法是以臨床、X線和病理檢查結果為主要依據,即經普通病理檢查發現病灶內有組織細胞浸潤即可確診。鑒於郎格罕細胞(LC)具有特殊的免疫表型和超微結構,國際組織細胞學會在1987年建議將此症確診的可信度分為三級,分級標準如下:
Ⅰ級(擬似診斷):具有典型的臨床表現,常規病理檢查發現郎格罕細胞(組織細胞)增殖浸潤。
Ⅱ級(提高診斷):在擬診基礎上,加以下四種染色有兩種或兩種以上陽性:
①ATP酶染色;
②S-100神經蛋白;
③α-D甘露糖苷酶染色;④花生凝集素。
Ⅲ級(確定診斷):光鏡檢查陽性,加電鏡下發現病變細胞內有Birbeck顆粒及(或)病變細胞的CD1a染色陽性。
提高診斷的可信度,首先有利於此症與其他類型的組織細胞增生症相鑑別,利於國際間診斷標準的統一,亦為加強國際間交流和進一步開展深入研究所必需。
鑑別診斷
本症應與某些骨骼、淋巴、肺部和皮膚器官的疾病,以及其他組織細胞增生症相鑒鑑別。
(一)與其他疾病的鑑別:
1.發熱、肝脾腫大、貧血:應與敗血症、傷寒、瘧疾、白血病、惡性組織細胞病、惡性腫瘤相鑑別。白血病的骨髓和外周血中可見白血病細胞。惡性組織細胞病的骨髓或病理活檢,可見分化不好的惡性組織細胞,皮疹多為出血點或瘤樣結節,肝脾大且多伴黃疸。
2.皮膚損害:本病的皮膚改變應與脂溢性皮炎、濕疹、膿皮病、血小板減少性紫癜或血管炎等相鑑別。皮膚念珠菌感染,可能與本病的鱗屑樣皮損相混淆,但本症皮損癒合後形成小的瘢痕和色素脫失為其特點,皮疹壓片可見分化較好的組織細胞。
3.肺部病變:常誤診為肺炎、粟粒性肺結核、肺含鐵血黃素沉著症,其無特徵性皮疹,無骨骼損害可作鑑別。肺部病變明顯的郎格罕細胞組織細胞增生症易與粟粒性肺結核混淆,鑑別要點在於:前者常有典型的出血性濕疹樣皮疹和骨質缺損損害,受累組織活檢及免疫組化見典型的組織上細胞,無結核接觸史、結核菌素試驗陰性、抗結核治療無效可排除粟粒型肺結核。後者常有結核接觸史,結核菌素試驗陽性,肝脾大較少見。
4.骨骼損害:上述骨骼的不規則破壞,軟組織腫脹、硬化和骨膜反應同樣見於骨髓炎、Ewing肉瘤、成骨肉瘤、神經母細胞瘤骨轉移、顱骨的表皮樣瘤以及纖維性發育不良等。惡性腫瘤骨轉移骨質損害有時誤診為郎格罕細胞組織細胞增生症,重點在惡性腫瘤多有原發腫瘤的明顯臨床表現,腫瘤活檢可確診。顱骨的溶骨性損害、突眼以及眼瞼瘀斑往往是神經母細胞瘤的表現。
與其他組織細胞增生症的鑑別:
1.竇性組織細胞增生症伴塊狀淋巴結腫大(Sinushistiocytosiswithmassivelymphadenopathy,SHML):SHML常表現為雙側頸淋巴結的無痛性腫大,除頸淋巴結受累外,余處淋巴結或結外病變(如皮膚、軟組織、骨損害)可見於40%以上的患者,皮膚病變常為黃色或黃色瘤樣,骨骼病變亦為溶骨性損害,X線表現很難與郎格罕細胞組織細胞增生症鑑別。SHML的組織學特點為組織細胞群的竇性增生,並與其他淋巴樣細胞和漿細胞相混合,病變細胞缺乏典型的郎格罕細胞核凹陷特點,且CD1a抗原陰性。超微結構檢查缺乏Birbeck顆粒,從而有別於郎格罕細胞。
2.噬血細胞性淋巴組織細胞增生症或家族性噬血細胞性淋巴組織細胞增生症(Familiahemophagocyticlymphohistiocytosis,FHL):是一組以發熱、全血細胞減少和肝脾腫大為特點的臨床綜述征,診斷的根據偏重於骨髓、淋巴結、肝脾和腦膜病變。高三酸甘油酯血症、低纖維蛋白原和腦脊液中淋巴細胞增多為本病的典型改變。FHL為常染色體隱性遺傳,診斷上有時與小兒繼發性噬血細胞綜合徵極難區別,後者亦稱病毒相關性噬血細胞綜合徵(Viralassociatedhemophagocyticsyndrome,VAHS)。VAHS又擴大套用於其他感染因素所誘發的類似綜合徵,甚至包括小嬰兒未接受任何免疫抑制劑治療或未有顯著感染的噬血細胞綜合徵病例。目前尚未缺乏實驗室或組織病理的方法,將這些綜合徵區別開來。如缺乏家族史,鑑別家族性或繼發性會相當困難。為此,組織細胞協會FHL研究組將FHL和VAHS統一命名為噬血細胞性淋巴組織細胞增生症(Hamophagocyticlymphohietiocytosis,HLH)。
3.郎格罕細胞組織細胞增生症(LCH)還應與惡性組織細胞病(惡組)、反應性組織細胞增多症(反應組)相鑑別,其鑑別要點見表
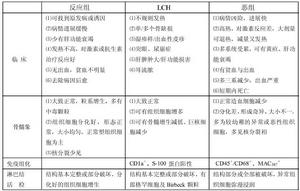 LCH與反應組、惡組的鑑別
LCH與反應組、惡組的鑑別治療
儘管LCH尚未完全證實屬於惡性腫瘤,但經驗證明以抗腫瘤化療藥物治療為主的治療方法,已使LCH的預後得到了明顯改變。除化療外,還對局部病灶作手術刮除或放療。由於LCH侵犯器官及病情輕重相差較大,治療應以人而異,分型論治。
化療
化療的目的是儘可能低劑量細胞毒藥物控制疾病,使器官功能損害減少至最低程度,但化療的強度要與疾病分級程度一致。對Ⅰ級、Ⅱ級者選用VP方案(VCR+Pred)化療6~8周,然後選用6MP(巰基嘌呤)和MTX(甲氨蝶呤)或單藥交替套用,療程1~2年。對Ⅲ級、Ⅳ級者選用VCP(VP+環磷醯胺)或VEP(VP+足葉乙甙)方案治療8~12周,病情好轉後改用6MP和MTX與之交替,療程2~3年。常用化療方案如下:
(一)VP方案:
VCR1.5mg/m2.次,QW,靜注;Pred60mg/m2.d,分次口服;兩藥聯用4~6周為一療程。(二)VCP方案:VCR和Pred同上;CTX(環磷醯胺):75mg/m2.次,d1~7、d15~21,口服或200mg/m2.次,靜滴,QW;三藥聯用4周為一療程。(三)EVP-Ⅰ方案:
E(Etoposide,VP16,足葉乙甙):100~150mg/m2.d,靜滴,d1~3,每3周連用3日;或用VM26(威猛,Teniposide,替尼泊甙);VCR:1.5mg/m2.次,QW,d1、d8、d15,靜注;Pred:60mg/m2.d,d1~21,分次口服;3周為一療程。用2~6個療程。
對Ⅲ級、Ⅳ級LCH患兒用EVP-Ⅰ方案治療2~6個療程後,6個月後每3個月一個療程,病情好轉達完全緩解(CR)後改為6MP與MTX兩藥聯合,以後交替輪迴治療至2~3年終止治療。
免疫治療
因本病與T細胞的免疫功能異常有關,對Ⅲ級、Ⅳ級病變者可於化療同時套用胸腺素(Thymosin),每日4mg,每日或隔日肌注,連用1個月後,病情穩定或好轉後,可改為每周2~3次,連用6個月。亦可使用α-干擾素治療。
局部治療
適用Ⅰ級、Ⅱ級病變,包括手術刮除病灶和放射治療。對初治的單一局部骨病變可單用病灶刮除術,對局部病變嚴重或持重負荷大的骨病變、復發病灶或多部位骨病變伴軟組織受累者、肺嗜酸細胞肉芽腫等,可聯合化療。病程在半年內的尿崩症亦可局部放療,總劑量600cGy,分次照射。對不宜作刮除術的骨骼病灶,也可用甲基強的松龍(Methylprednisolone)作局部病灶內注射,每次75~150mg。
對LCH局部皮膚病變,可局部套用皮質激素或氮芥,對全身瀰漫性皮膚病變採用化療也可使皮膚損害得到控制。
其他治療
(一)LCH伴尿崩症時用DDAVP(Desmopressin去氨加壓素,Minirin彌凝),每次5~10μg滴鼻或噴鼻,每日1~2次;或口服Minirin片50~100µg,每日3次。
(二)新近報告用環孢黴素A(CyclosporineA)對器官功能損害的完全恢復和LCH的病損的消退多數是有效的。環孢黴素抑制細胞因子介導(cytokine-mediated)的細胞活化作用,被認為其對LCH的發病中起重要作用。治療LCH有一定療效,用法與劑量:6~12mg/kg,,分2~3次口服,連服用6~12個月(或CSA+VP16、Pred)。
(三)α-干擾素(α-Interferon,α-IFN):每日100~150萬IU,肌注,連用10周,以後每周3日,共用14個月,並與強的松並用。
(四)2-氯脫氧腺苷(2-chlorodeoxyadenosine,2CDA):5~6.5mg/m2.d,連續3天為1個療程,每3~4周重複1個療程,共4~6個療程。
(五)噴司他丁(Pentostatin,2-Deoxycofarmycin):本品是一種腺苷脫氨酶的強抑制劑,有報導用於治療難治性多系統性LCH。
(六)LCH如主要骨損害難愈,可用消炎痛(Indomethacin),因LCH的骨病變部位PGE2水平增高而破壞骨骼,用消炎痛可抑制PGE2水平的增高,達到減少骨的破壞。用法:消炎痛12.5mg/kg.d,分2~3次口服,用6周為一療程,如有效再用2~6周,如無效,則停藥。副作用:有頭痛,消化道出血,偶有血細胞減少。
(七)Pamidronate(帕米磷酸鈉,丙氨膦酸鈉)(30mg/支、250mg/片)、Bonefone(Disodiumclodronate,氯曲膦酸鈉,骨膦)(400mg/粒)為破骨細胞性骨溶解抑制劑,抑制破骨細胞活性而抑制骨吸收作用。
(八)對Ⅳ患兒,有條件者採用異基因骨髓移植。(九)對繼發侏儒的患兒可試用生長激素。(十)加強支持療法,積極預防和控制感染。
治療標準
根據國際組織細胞學會1991年制定的LCH-1治療方案,將療效標準確定如下。
(一)痊癒:症狀和(或)客觀檢查徵象完全消失。
(二)好轉:症狀和(或)客觀檢查徵象消退,無新病灶出現。
(三)穩定:指症狀和(或)客觀徵象持續存在,但無新病灶出現。
(四)進展或惡化:症狀和(或)客觀檢查徵象較確診時有進展和(或)出現新病灶或舊病灶復發。
(五)混合性治療反應:指原有病灶好轉或消失,卻又有新的病灶出現者
預後
根據不同的類型和分度(級),其預後差異很大,發病年齡越小(如小於2歲),受累器官越多,器官功能障礙越明顯則預後越差。採用聯合化療後,長期存活率65%左右。部分患兒可出現一些遠期後遺症,如肺功能不全、肝纖維化、脂肪肝、尿崩症、生長遲緩、性發育不良等,也可有中樞神經系統功能損害的表現。
