Marcus理論
套用該理論在無機化學、有機化學學科及其他領域處理了眾多電子轉移體系並獲得了令人滿意的結果。由於該理論的重大貢獻Marcus榮獲了1992年諾貝爾化學獎。
電子轉移反應是當電子給體與受體由擴散而靠近達到一定距離時,因二者存在電勢層,電子便從給體轉移給受體。最簡單的電子轉移(單電子轉移)反應如同位素交換反應:
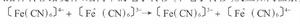 舉例
舉例式中*表示放射性同位素。
由反應看出,電子轉移僅發生在價電子層上。由於反應物與產物未發生質變,所以此反應的標準自由能變化為零。通過測定反應速率常數即可確定控制該反應速率的因素。當電子轉移反應進行時,反應速率是隨離子與配體間距離的變化、離子的結構變化、離子同溶劑分子相連線構成雙極子的結構變化等因素而變化的。如上述反應在水溶液中Fe —HO的距離比Fe —HO的距離大1.4×10 ,此鍵鍵長的變化引起33kJ/mol反應自由能勢壘的增高(即反應活化能增大),而且由於向Fe 配位的溶劑分子轉向電荷大的Fe 配位的溶劑分子井然有序,此溶劑分子的配位能差即反應自由能勢壘超出33kJ/mol。與此同時,由於反應坐標的起伏波動,反應物(兩個離子)的結構與生成物(兩個離子)的結構必須相近。
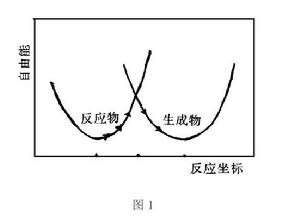
對於如上單電子轉移反應,應考慮反應物離子振動模式和同反應物離子相配位的溶劑分子的結構以及對極化作用較小的溶劑分子的振動模式等的反應坐標,對通常的電子轉移反應必須考慮反應物、生成物及其所處的溶劑環境,從多維勢能面綜合考慮。反應物體系與生成物體系的勢能面UR與UP的一維曲線如圖所示。由於反應體系與生成體系反應熱的起伏波動,當達到兩面相交的能量狀態(活化過渡態)處,因距離足夠小,兩反應物間相互作用非常強,反應物經較低的勢能面翻躍馬鞍形能峰達到生成物的幾率為1時(按箭頭所示),則發生電子轉移反應。
 電子轉移引發反應
電子轉移引發反應根據Marcus理論,電子轉移反應的活化自由能變化(反應能峰)可由下式表述:
ΔG =(λ/4)(1+ΔG/λ) 。 (1)
式中λ稱為反應重組能,是鍵能與溶劑分子配位能之和,將由此得出的電子轉移反應的活化自由能變化代入反應速率的過渡狀態理論公式:
ΔG =-RTlnk +c。 (2)
k≠是反應速率常數。由此可計算出反應物達到過渡狀態的幾率,而反應速率是由達到過渡狀態的幾率和在過渡狀態區域由反應物向生成物轉移的幾率之乘積來表示。
發展前沿-單電子轉移反應
單電子轉移反應在無機化學中是很重要的,而以往我們所討論的大多數有機自由基反應主要是鍵的均裂、奪取原子及在反應的引發和增長階段中的加成、重排、結合、歧化等各步驟的結合。實際上在一個有機分子中加上或除去一個電子而引發的反應也是很重要的且並不少見。
從廣泛的意義上講,反應若按單電子轉移途徑進行,則是自由基和離子型反應的結合,反應產生一類自由基型中間體。反之,若按極性途徑進行,則發生雙電子轉移,反應不經過自由基中間體而產生新的化學鍵。因此人們可以通過檢測自由基中間體及分析相關產物來區別這兩種不同的反應途徑。由於順磁共振技術的發展,已使人們可以較好地對自由基反應過程進行觀察並能找到明確的證據。
單電子轉移反應可以在化學試劑和光的作用及電化學條件下產生。許多無機試劑都表現出很強的單電子氧化性,它們與有機分子的許多反應是通過單電子氧化形成的自由基型中問體進行的。醯基過氧化物和醇金屬試劑,碳陰離子(基),硫化物等也都易引發單電子轉移反應。許多過渡金屬離子具有不止一個比較穩定的氧化態。所以,過渡金屬離子在涉及單電子轉移的過程中經常被用作催化劑和試劑。
