
內容
單細胞全基因組測序 技術是在單細胞水平對全基因組進行擴增與測序的一項新技術。其原理是將分離的單個細胞的微量全基因組DNA進行擴增,獲得高覆蓋率的完整的基因組後進行高通量測序用於揭示細胞群體差異和細胞進化關係。
從方法學角度來看,獲得高覆蓋率高保真性的全基因組擴增產物是準確全面的測序結果的保障。多重置換擴增(multiple displacement amplification,MDA)利用隨機引物和等溫擴增可以獲得高保真的DNA大片段,但該方法的主要缺陷在於非平衡的基因組覆蓋率、擴增偏倚、嵌合序列及非特異擴增等 。儘管各種改進的策略正在逐步減少這些缺陷,高覆蓋率、高保真性及高特異性的擴增仍然是亟待解決的問題。另外,還有科研人員利用DOP-PCR進行全基因組擴增(whole-genomeamplification,WGA)及DNA測序對單個乳腺癌細胞進行了拷貝數變異的分析,進而推斷出細胞的群體結構和腫瘤的進化過程。但是由於該方法的基因覆蓋率較低,而且不能在單個核苷酸的解析度上評價單個腫瘤細胞的遺傳學特徵,故並不能檢測在腫瘤發展過程中發揮重要作用的單個核苷酸的改變。2012年,哈佛大學謝曉亮院士在《Science》發表了單細胞全基因組擴增新技術 MALBAC(Multiple Annealing and Looping Based Amplification Cycles,簡稱MALBAC),即多次退火環狀循環擴增技術 。不同於以往的非線性或指數型擴增方法,MALBAC技術利用特殊引物,使得擴增子的結尾互補而成環,從而很大程度上防止了DNA的指數性擴增,從而解決了基因組擴增對微量初始模板過大的擴增偏倚,並使基因組測序的模板需求量從µg級降至單細胞水平。MALBAC技術原理如下:
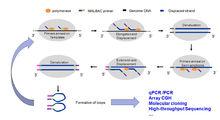 圖1:MALBAC技術原理
圖1:MALBAC技術原理圖1:MALBAC技術原理
MALBAC Primers having a 27-ntcommon sequence followed by eight random nucleotides are annealed to thegenomic DNA template. Strand-displacement synthesis generates partialamplicons, which are subsequently denatured from the template at 94°C. Primingto new positions on the genomic DNA template generates more partial amplicons,which increases coverage of the genome with a resulting reduction inamplification bias. Priming nad extension on the partial amplicons yield completeamplicons having the MALBAC primer sequence at 5’ end and its complementarysequence at the 3’ end. Denaturation at 94°C regenerates the original templateand a now larger and more diverse pool of partial amplicoms. Full ampliconsform loops, which may be resistant to subsequent amplification andhybirdization. Full amplicons are generated for eight cycles and theexponentially amplified by about 14-21 cycles using primers complementary tothe common region of the MALBAC primers .
常見的全基因組擴增方法(whole-genomeamplification,WGA)比較如下:
表1:常見WGA方法比較
 表1:常見WGA方法比較
表1:常見WGA方法比較通過比較我們發現, MALBAC技術具有如下優勢:
降低PCR擴增偏倚,使得單細胞中93%的基因組能夠被測序 。
這種方法使得檢測單細胞中較小的DNA序列變異變得更容易,因此能夠發現個別細胞之間的遺傳差異。這樣的差異可以幫助解釋癌症惡化的機制,生殖細胞形成機制,甚至是個別神經元的差異機制。
靈敏度高:單細胞、單染色體或0.5pg的基因組DNA即可進行擴增。
擴增均勻性顯著優於其它技術,測序數據可進行CNV分析,用於21三體等染色體變異檢測。
擴增產物用途廣泛:可用於二代測序、微陣列、qPCR、基因克隆。
1.降低PCR擴增偏倚,使得單細胞中93%的基因組能夠被測序 。
這種方法使得檢測單細胞中較小的DNA序列變異變得更容易,因此能夠發現個別細胞之間的遺傳差異。這樣的差異可以幫助解釋癌症惡化的機制,生殖細胞形成機制,甚至是個別神經元的差異機制。
2.靈敏度高:單細胞、單染色體或0.5pg的基因組DNA即可進行擴增。
3.擴增均勻性顯著優於其它技術,測序數據可進行CNV分析,用於21三體等染色體變異檢測。
4.擴增產物用途廣泛:可用於二代測序、微陣列、qPCR、基因克隆。
目前,MALBAC技術現在已經成功套用於人類單精子、植入前產前篩查的囊胚和極體、早期發育胚胎、腫瘤細胞、刑偵現場痕量物證和部分微生物。
參考文獻
Yilmaz S, Singh AK.Single cell genome sequencing. Curr Opin Biotechnol 2012; 23(3): 437-43.
Zong CZ, Lu SJ,Chapman AR,XieXS.Genome-Wide Detection of Single Nucleotideand CopyNumber Variations of a Single Human Cell. Science 2012;338(6114):1622-6.
Lu SJ, Zong CZ, Xie XS, et al. ProbingMeiotic Recombination and Aneuploidy of Single Sperm Cells by Whole GenomeSequencing using MALBAC. Science 2012;338(6114):1627-30.
Balogh M K. Application of whole genomeamplification for forensic analysis. Elsevier 2006; 1288:725-727.
Pinard R, de Winter A, Sarkis G J, etal. Assessment of whole genome amplification-induced bias throughhigh-throughput, massively parallel whole genome sequencing. Bmc Genomics 2006;7(1): 216-20.
1.Yilmaz S, Singh AK.Single cell genome sequencing. Curr Opin Biotechnol 2012; 23(3): 437-43.
2.Zong CZ, Lu SJ,Chapman AR,XieXS.Genome-Wide Detection of Single Nucleotideand CopyNumber Variations of a Single Human Cell. Science 2012;338(6114):1622-6.
3.Lu SJ, Zong CZ, Xie XS, et al. ProbingMeiotic Recombination and Aneuploidy of Single Sperm Cells by Whole GenomeSequencing using MALBAC. Science 2012;338(6114):1627-30.
4.Balogh M K. Application of whole genomeamplification for forensic analysis. Elsevier 2006; 1288:725-727.
5.Pinard R, de Winter A, Sarkis G J, etal. Assessment of whole genome amplification-induced bias throughhigh-throughput, massively parallel whole genome sequencing. Bmc Genomics 2006;7(1): 216-20.
