基本信息
另外,其用戶圖形化界面(GUI)工具為AutoDockTools(ADT),其第二代產品為AutoDock Vina。
AutoDock包含但不局限於以下套用:
•X-射線晶體學
•基於結構的藥物設計
•先導化合物最佳化
•虛擬篩選
•組合庫設計
•蛋白—蛋白對接
•化學機制研究
開發這一程式的靈感源於設計生物活性化合物中遇到的問題,特別是計算機輔助藥物設計領域。生物大分子的 X-射線衍射技術的進步為我們提供了更多重要的蛋白和核酸分子的結構。這些結構可以作為生物活性物質的靶標,用於控制動植物的疾病,或者可以使人們簡單的理解活性物質在生物學方面的作用機理。準確的了解蛋白靶標和這些活性小分子之間的相互作用是十分重要的。因此,開發者的目標就是為科研工作者提供一個計算工具,幫助他們研究生物大分子(蛋白質)與小分子(配體)複合物的相互作用。
工作原理
任何對接計算都有兩個相互矛盾的方面需要平衡:在儘可能精準的計算與合理(有限)的計算資源之間達到一個平衡。理想的步驟是通過搜尋整個系統可能的自由度,在底物和目標蛋白的結合能中找到全局能量極小值。然而這樣的工作只能在大型的工作站上實現,並且耗費和結構生物學家進行晶體結構修飾相當的時間。為了解決這一問題,很多對接軟體簡化了對接的步驟。Autodock 通過兩種方法的結合使用解決了以上的問題:快速的基於格點能量的計算方法(rapid grid-based energy evaluation)和有效的扭轉自由度搜尋方法(efficient search of torisional freedom)。
AutoDock軟體由 AutoGrid 和 AutoDock兩個程式組成。其中 AutoGrid 主要負責格點中相關能量的計算,而 AutoDock 則負責構象搜尋及評價。
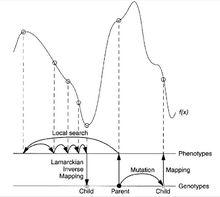 LGA算法操作過程圖
LGA算法操作過程圖AutoDock 在早期版本中使用的是模擬退火算法(Simulated Annealing Algorithm)來尋找配體與受體最佳的結合位置狀態,而從 3.0 版本開始使用一種改良的遺傳算法,即拉馬克遺傳算法(Lamarckian Genetic Algorithm,LGA)。測試結果表明,LGA 比傳統的遺傳算法和模擬退火具有更高的效率。在 LGA 方法中,作者把遺傳算法和局部搜尋(Local search)結合在一起,遺傳算法用於全局搜尋,而局部搜尋用於能量最佳化。LGA 算法引入了拉馬克的遺傳理論,這個操作過程右圖。
同時在AutoDock中配體和受體之間結合能力採用能量匹配來評價。在1.0和2.0版本中,能量匹配得分採用簡單的基於AMBER力場的非鍵相互作用能。非鍵相互作用來自於三部分的貢獻:范德華相互作用,氫鍵相互作用以及靜電相互作用。而在3.0之後的版本中AutoDock提供了半經驗的自由能計算方法來評價受體和配體之間的能量匹配。
為了加快計算速度,AutoDock 採用格點對接的方法,但與 DOCK中格點對接的處理方法有明顯的區別。DOCK 中,格點上保存的不是能量,而是僅與受體有關的特徵量。而在 AutoDock 中,格點上保存的是探針原子和受體之間的相互作用能。
對於范德華相互作用的計算, 每個格點上保存的范德華能量的值的數目與要對接的配體上的原子類型(表 3)的數目一樣。如果一個配件中含有 C、O 和 H 三種原子類型,那么在每個格點上就需要用三個探針原子來計算探針原子與受體之間的范德華相互作用值。當配體和受體進行分子對接時,配體中某個原子和受體之間的相互作用能通過周圍 8 個格點上的這種原子類型為探針的格點值用內插法得到。
| 原子類型 | 原子類型 | ||
| H | Non H-bonding Hydrogen | S | Non H-bonding Sulphur |
| HD* | Donor 1 H-bond Hydrogen | Cl | Non H-bonding Chlorine |
| HS | Donor S Spherical Hydrogen | CL | Non H-bonding Chlorine |
| C* | Non H-bonding Aliphatic carbon | Ca | Non H-bonding Calcium |
| A* | Non H-bonding Aromatic Carbon | CA | Non H-bonding Calcium |
| N* | Non H-bonding Nitrogen | Mn | Non H-bonding Manganese |
| NA* | Acceptor 1 H-bond Nitrogen | MN | Non H-bonding Manganese |
| NS | Acceptor S Spherical Nitrogen | Fe | Non H-bonding Iron |
| OA* | Acceptor 2 H-bonds Oxygen | FE | Non H-bonding Iron |
| OS | Acceptor S Spherical Oxygen | Zn | Non H-bonding Zinc |
| F | Non H-bonding Fluorine | ZN | Non H-bonding Zinc |
| Mg | Non H-bonding Magnesium | Br | Non H-bonding Bromine |
| MG | Non H-bonding Magnesium | BR | Non H-bonding Bromine |
| P | Non H-bonding Phosphorus | I | Non H-bonding Iodine |
| SA* | Acceptor 2 H-bonds Sulphur |
靜電相互作用的計算採用了一個靜電勢格點,在格點上儲存受體分子的靜電勢。當配體和受體分子對接時,某個原子和受體之間的靜電相互作用能通過周圍格點上靜電勢以及原子上的部分電荷就可以計算得到。
計算氫鍵相互作用時,格點的處理和范德華相互作用有點類似,每個格點上需要保存配體分子中所有氫鍵給體與氫鍵受體之間的相互作用能量,而且這些能量都是在氫鍵在最佳情況下的氫鍵能量值。
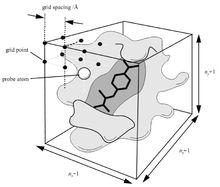 AutoDock格點對接示意圖
AutoDock格點對接示意圖以上格點能量的計算都是由 AutoDock 中的 AutoGrid 程式計算得出的,AutoDock 格點對接示意圖如左圖所示。AutoDock 格點對接的基本流程如下:首先,用圍繞受體活性位點的胺基酸殘基形成一個範圍更大的 Box,然後用不同類型的原子作為探針(probe)進行掃描,計算格點能量,此部分任務由 AutoGrid 程式完成。然後 AutoDock 程式對配體在 Box 範圍內進行構象搜尋(conformational search),最後根據配體的不同構象(conformation),方向(orientation)、位置(position)及能量(energy)進行評分(scoring),最後對結果進行排序(ranking)。
相關程式
AutoDockTools
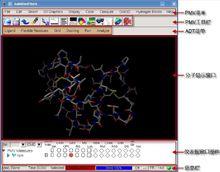 ADT1.5主界面及視窗部件
ADT1.5主界面及視窗部件AutoDock 本身所包含的 AutoDock 以及 AutoGrid 程式是完全在命令符下操作的軟體,沒有用戶圖形化界面,但是如果使用 AutoDockTools 程式(以下簡稱ADT),就可以在幾乎完全圖形化的界面中完成分子對接以及結果分析等工作。ADT是Scripps研究所的Molecular Graphics Laboratory (MGL)在 Python Molecular Viewer(簡稱 PMV,Python 語言開發)基礎上開發的針對AutoGrid和 AutoDock程式開發的圖形化的分子可視化及對接輔助軟體,目前最新版本為 1.5.2。在這裡我們使用的版本為 1.5.1,它的主界面主要包含以下幾個部分:
•PMV 選單:主要通過使用選單命令對分子進行相關的操作,以及進行可視化設定
•PMV 工具列:PMV 選單中一些常用命令的快捷按鈕
•ADT 選單:AutoGrid 和 AutoDock 的圖形化操作選單
•分子顯示視窗:3D 模型分子的顯示和操作視窗
•儀錶板視窗部件:快速查看及設定分子的顯示模型以及著色方式
•信息欄:顯示相關操作信息
AutoDock Vina
AutoDock Vina也是一款由MGL實驗室開發的分子對接軟體。與AutoDock 4.0相比,AutoDock Vina提高了結合模式預測的平均準確度,通過使用更簡單的打分函式加快了搜尋速度,並且在處理約20個可旋轉鍵的體系時仍然能提供重現性較好的對接結果。
PyRx
AutoDock 當前只能實現單個配體和受體分子之間的對接,程式本身還沒有提供虛擬篩選功能(Virtual Screening)。目前,可以使用 Linux/Unix 中的 Shell 以及 Python 語言實現此功能,但對於不熟悉Linux/Unix系統的研究人員來說,可以嘗試利用PyRx進行虛擬篩選工作。
PyRx是基於AutoDock(4.0及以上版本)或者AutoDock Vina的虛擬篩選工具。它具有簡單易用的對接外掛程式,使其成為計算機輔助藥物設計的有力工具;它也包含合理藥物設計所必需的表單功能和可視化引擎。
