
1.簡介
顯微光學成像,通常也稱“光學顯微成像”,或“光學顯微術”(Optical Microscopy,或Light Microscopy),是指透過樣品或從樣品反射回來的可見光,通過一個或多個透鏡後,能夠得到微小樣品的放大圖像的技術。所得圖像可以通過目鏡直接用眼睛觀察,也可以用感光板或數位化圖像探測器如CCD、CMOS進行記錄,還可以在計算機上進行顯示和分析處理。
採用明場照明方式的普通光學顯微術通常存在三個方面的局限性:一是只能對深色樣品(透射型)或強反光樣品(反射型)進行成像;二是光學衍射極限限制了該技術的最高解析度約為200 nm;三是離焦信息會降低圖像對比度。基於樣品中(外源或內源)螢光分子的激發和螢光發射的螢光顯微術(Fluorescence Microscopy),可以突破無法對透明樣品成像的局限,但解析度受限和離焦干擾的問題仍需要採取其他措施才能加以解決。
2 無色透明樣品的顯微光學成像
對於無色透明樣品,例如未經任何處理的活細胞,採用普通的光學顯微術一般會由於缺乏足夠的對比度而無法直接觀察和成像,原因是這些樣品的內部結構並不會導致照明光強的吸收,從而不能形成有效的圖像襯度。通常的做法是利用一些特異性染料來標記樣品中的不同結構以提高圖像對比度,但是染色過程往往會改變甚至破壞樣品,從而難以得到樣品本身的真實信息。然而,採用一些改進的技術,可以使該局限性得到一定程度的改善。這種技術大致可分成兩類,一類是通過改變照明方式來實現,另一類則是通過將樣品對照明光相位的改變數轉變為光強分布來實現,後者通常稱為“相襯法”。
一、照明方式:
1.明場照明:
明場照明是光學顯微術中最簡單的一種方式。在倒置顯微鏡中,只需採用白光從樣品下方照射,在樣品上方觀察透射光即可,圖像對比度取決於樣品不同部位對光的吸收。採用這種方式最大的缺點是大多數生物樣品的對比度都很低,並且會由於離焦信息的干擾導致解析度也較低。但優點是設備簡單,樣品也無需進行繁瑣的處理。
2.斜照明:
相比明場照明,斜照明方式的優點是能夠使樣品的圖像產生一種三維(3D)的效果,從而凸顯出樣品中一些原本看不到的結構。目前常用的“霍爾曼調製對比度”技術就是基於斜照明方式發展起來的,一般用在細胞培養室中使用的倒置顯微鏡上。但採用斜照明方式並不能顯著提高對比度和解析度。
3.暗場照明:
採用暗場照明方式的暗場顯微術通過採集樣品散射的光線來提高無色透明樣品的圖像對比度。為了只收集樣品的散射光,暗場照明方式需要採用準直光源使進入像平面的透射光(未散射的部分)強度降到最低。相比另外兩種方式,採用這種照明方式的技術其缺點主要在於光強較弱導致的成像時間過長。
4.萊因伯格照明
萊因伯格照明(Rheinberg Illumination)方式實際上是暗場照明的一種特殊變體。其主要區別通過在聚光器之前插入彩色的透明濾光片,從而可以用顏色來區分樣品中不同空間的成分,例如可將有結構的樣品顯示為紅色,而均勻的背景則顯示為藍色,或者其它不同顏色的組合(效果也會有所不同)。
5.其它照明方式
如正交偏振光照明,其襯度取決於樣品不同部位對偏振光的不同旋轉程度。
 顯微光學成像
顯微光學成像圖1.相同樣品(鏡頭紙)在明場(左)和暗場(右)照明方式下的成像效果比較
二、相襯法:
光學顯微術對無色透明樣品無法直接成像的原因在於樣品不改變照明光的強度分布,但實際上這些樣品由於厚度和折射率的差異會改變照明光場的相位分布,只是由於人眼和探測器僅對強度敏感,不能直接記錄相位信息。因此,假如能把照明光場經過樣品後的相位改變數的分布轉化成光強分布,就能夠實現對這種樣品(通常稱為“相位”樣品)的成像,這種技術稱為“相襯法”,用於光學顯微術中則形成了各種“相襯顯微術”。
1.澤尼克相襯顯微術
20世紀30年代,荷蘭物理學家澤尼克(Zernike)首次提出了相襯技術(1953年獲得諾貝爾獎),其原理是通過將直射光(即零頻光)的相位改變±90°(即1/4波長的光程差)並適當衰減,從而使直射光和衍射光發生干涉而使像平面上的復振幅分布近似正比於物體的相位分布,將“看不見”的相位變化轉化為“可見”的強度分布。在具體光路上,澤尼克相襯顯微鏡需要一個能夠產生錐形照明光的圓環型聚光器,以及位於物鏡後焦面處的一個對應於該錐形照明光通過區域的相位環。採用該技術,可以方便地實現對無染色的活細胞樣品的直接觀察和成像,但其缺點是不適合厚樣品(上述近似關係不成立)和極微小樣品(光暈現象嚴重)。
2.微分干涉相襯顯微術
微分干涉相襯(Differential Interference Contrast, DIC)顯微術是目前一些較高端的倒置顯微鏡中通常會配備的另一種相襯技術,但相比基於澤尼克法,這種技術實現起來要相對複雜一些,也更昂貴一些。在DIC系統中,需採用兩個特殊稜鏡,稱為渥拉斯頓稜鏡,其中一個裝在聚光器內,作用是將照明光分成彼此錯開的尋常光和非尋常光,並使其光程差小於物鏡的最高解析度;另一個裝在物鏡的後焦面處,使經過樣品後的兩束光重新合併成一束並在像面發生干涉。在樣品中折射率發生突變的地方,兩束光干涉的結果會使圖像產生“浮雕”的立體效果。該技術要求採用偏振光,因此光路中還包含兩個偏振器,一個位於聚光器前(起偏器),另一個位於物鏡後(檢偏器)。
3.干涉反射顯微術:
干涉反射顯微術(Interference Reflection Microscopy)是運用干涉原理的另一種相襯顯微術,也稱為“反射干涉相襯(Reflected Interference Contrast, RIC)顯微術”。RIC主要用於檢測是否存在粘附,如細胞是否很好貼壁等。RIC系統和DIC系統的不同之處,除了探測的是反射光而不是透射光之外,所採用的稜鏡也有所不同。
3 螢光顯微術
利用螢光分子標記細胞樣品中某些特定結構或成分,用波長較短的光激發這些螢光分子使其處於高能態,再對其退激過程中所發射的波長相對較長的螢光進行顯微成像,即為螢光顯微術。採用該技術,不僅可以克服普通光學顯微術難以直接對無色透明的細胞樣品進行成像的缺點,更重要的是還能通過標記細胞中特定的結構或者分子、離子實現“功能成像”。由於離焦干擾等原因,普通寬場螢光顯微術解析度並不高,但利用一些特殊技術可以顯著提高解析度和對比度。
1.共聚焦雷射掃描顯微術
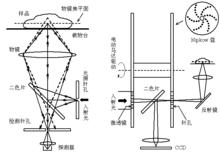
共聚焦雷射掃描顯微術(Confocal Laser Scanning Microscopy, CLSM)的基本原理是利用一對共軛針孔進行“空間濾波”,將焦平面以外的雜散光濾除以提高縱向解析度,實現非侵入式的“光學切片”。CLSM的橫向解析度約為100-200 nm,縱向解析度可達500 nm,大大優於普通非共焦方式的螢光顯微術;同時共焦針孔的套用也極大地提高了圖像信噪比。這些優點使CLSM被廣泛套用於細胞生物學的研究,尤其是亞細胞層次空間結構和功能的研究。
然而CLSM採用的是逐點掃描的成像方式,成像速度受到限制。近年來發展起來的轉盤式共聚焦顯微術(Spinning Disk Confocal Microscopy, SDCM)仍利用CLSM的共焦針孔原理,但由於採用高速旋轉的轉盤實現了非常多焦點的近似面照明,所得螢光可以採用CCD採集,在保持圖像質量達到傳統CLSM水平的同時成像速度可達視頻要求,成為活細胞內單分子螢光成像的有力工具。
 顯微光學成像
顯微光學成像圖2.共聚焦雷射顯微術(左)和轉盤式共聚焦顯微術(右)原理示意圖
2.全內反射螢光顯微術
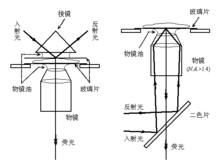
當一束光從光密介質射向光疏介質時,並且入射角大於臨界角時,將發生全內反射(Total Internal Reflection, TIR)。由於光的波動效應,此時仍會有少量光能量穿透界面並沿著平行界面的方向傳播,稱為隱失波(Evanescent Wave)全內反射螢光顯微術(TIR Fluorescence Microscopy, TIRFM)就是利用這種隱失波來激發樣品螢光的,由於其穿透深度一般只有100-200 nm,因而縱向解析度和圖像信噪比均大大提高。常見的TIRFM有稜鏡型和物鏡型兩類。TIRFM一般套用於研究細胞膜或膜內附近區域的動態過程,如信號轉導等。
 顯微光學成像
顯微光學成像圖3.稜鏡型(左)和物鏡型(右)全內反射螢光顯微術示意圖
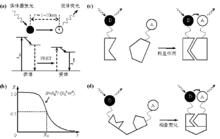
3.螢光共振能量轉移
螢光共振能量轉移(Fluorescence Resonance Energy Transfer, FRET)是指兩個不同螢光分子(或螢光團)之間發生的非輻射能量轉移。提供能量與接受能量的螢光分子分別稱為供體(Donor)和受體(Acceptor),FRET效應強烈依賴於兩者之間的距離(不超過10 nm),同時要求供體發射光譜與受體激發光譜有一定重疊,此時若激發供體,供體會通過FRET效應將能量轉移給受體,導致供體的螢光強度比其單獨存在時要低得多,而受體螢光卻大大增強。利用該效應進行成像,可實現非常高的空間解析度(nm量級),從而突破光學衍射極限的限制。利用FRET技術即可以研究分子之間的相互作用,也可以研究單分子內兩亞基之間的結合或分子構象的改變等。在實際套用中,FRET技術結合前面提到的SDCM、TIRFM等技術,可用於活細胞內單分子螢光成像的研究。
 顯微光學成像
顯微光學成像圖4. 分子間(上)和分子內(下)螢光共振能量轉移原理示意圖
4 超解析度技術
上述幾種技術除了FRET可以從物理機制上突破光學衍射極限外,其他技術的解析度仍然是受其限制的。近十年來,科學家們又發展了多種能夠一定程度上突破該限制的新技術,統稱為“超分辨技術”,主要有三類:結構光照明顯微術(Structured Illumination Microscopy, SIM)、受激輻射耗盡顯微術(Stimulated Emission Depletion Microscopy, STED)和單分子定位和構圖技術(Single Molecule Localization and Composition),其中單分子定位技術又包括光激活定位顯微術(photoactivated localization microscopy, PALM)和隨機光學重建顯微術(stochastic optical reconstruction microscopy, STORM)兩種。採用這些超解析度技術之後,成像效果相比傳統的寬場螢光顯微術甚至CLSM和TIRFM等均有顯著提高,目前文獻報導的橫向解析度已經達到20-30 nm左右。
 顯微光學成像
顯微光學成像圖5.超解析度顯微術和傳統高解析度顯微術的成像效果比較
擴展閱讀
[1] Paddock SW. Mol. Biotechnol., 2000, 16: 127
[2] Ichihara A, Tanaami T, Isozaki K et al. Bioimages, 1996, 4: 57
[3] Adams MC, Salmon WC, Gupton SL et al. Methods, 2003, 29: 29
[4] Ovechkina Y, Maddox P, Oakley CE et al. Mol. Biol. Cell, 2003, 14: 2192
[5] Hermann M, Pirkebner D, Draxl A et al. Transplant. Proc., 2005, 37: 3409
[6] Tadakuma H, Yamaguchi J, Ishihama Y et al. Biochem. Biophys. Res. Commun., 2001, 287: 323
[7] Tadakuma H, Ishihama Y, Toshiharu S et al. Biochem. Biophys. Res. Commun., 2006, 344: 772
[8] Iino R, Koyama I, Kusumi A. Biophys. J., 2001, 80: 2667
[9] Mashanov GI, Tacon D, Knight AE et al. Methods, 2003, 29: 142
[10] Mashanov GI, Tacon D, Peckham M et al. J. Biol. Chem., 2004, 279: 15274
[11] Sako Y, Minoghchi S, Yanagida T. Nat. Cell Biol., 2000, 2: 168
[12] Murakoshi H, Iino R, Kobayashi T et al. Proc. Natl. Acad. Sci. USA, 2004, 101: 7317
[13] Duncan RR. Biochem. Soc. Trans., 2006, 34: 679
[14] Wang ZF, Shah JV, Berns MW et al. Biophys. J., 2006, 91: 343
[15] Leake MC, Chandler JH, Wadhams GH et al. Nature, 2006, 443: 355
[16] Douglass AD, Vale RD. Cell, 2005, 121: 937
[17] Gorelik J, Shevchuk A, Ramalho M et al. Proc. Natl. Acad. Sci. USA, 2002, 99: 16018
[18] 林丹櫻, 馬萬雲. 活細胞內的單分子螢光成像方法, 物理, 2007, 36(10): 783-790.
[19] Schermelleh L, Heintzmann R and Leonhardt H. J. Cell Biol., 2010, 190: 165.
