芳香化酶(aromatase , AR) (也叫雌激素合成酶) )是微粒體細胞色素P450 的一種複合酶, 它由血紅蛋白P450和還原型輔酶NADPH 組成, 廣泛存在於卵巢、肝臟、肌肉、脂肪和乳腺癌細胞中,是催化生物體內雄激素向雌激素轉化的關鍵酶和限速酶,可將雄激素的A環芳香化, 脫去19 位的碳原子並將1位的羰基轉化為羥基, 這樣就將催化雄烯二酮和睪酮等雄激素轉化為雌酮和雌二醇, 後兩者即為絕經後女性雌激素的主要來源。雌激素與腫瘤進展有關,芳香化酶在雌激素生物合成中起最終的限速催化作用。
芳香化酶抑制劑(aromataseinhibitor,AI)能特異性導致芳香化酶失活, 阻斷芳構化反應, 抑制雌激素生成,降低血液中雌激素水平從而達到治療乳腺癌的目的。多用於抗雌激素(他莫昔芬)治療失敗的絕經後晚期乳腺癌患者。常用的芳香化酶抑制劑:依西美坦、來曲唑、阿那曲唑。
1.芳香化酶的分布與調節
雌激素可在男性和女性的各種組織中合成。絕經後婦女循環雌激素水平主要依靠脂肪組織合成的雌激素來維持, 然而人們發現絕經後婦女乳腺組織的雌二醇水平比血漿中高10倍。有報導指出, 芳香酶的活性以及芳香化酶的mRNA在正常乳腺組織和乳腺腫瘤中都存在。某些臨床觀察顯示: 腫瘤內的芳香化與腫瘤對芳香化酶抑制劑治療後雌激素合成受抑的反應相關, 局部雌激素的產生對腫瘤的增生可能也有重要作用。然而, 在人體乳腺癌組織勻漿中測得的芳香化酶活性相對較低, 而且不足以催化形成足量的雌激素去激活雌激素受體。另有研究表明, 局部雌激素濃度足以刺激腫瘤生長。同時組織培養發現, 某些由雌激素刺激的腫瘤也可以通過睪丸酮加速增生, 且該刺激作用可以通過芳香化酶抑制劑阻滯。提示睪丸酮經芳香化生成雌激素。由此可見, 腫瘤中的芳香化酶對刺激腫瘤增生的雌激素的生成具有重要意義。
2. 芳香化酶抑制劑的分類
研究表明:有三分之二的乳腺癌是雌激素依賴性的, 它們能在雌激素減少後得以消退。1973年, Schwarsel 首次提出芳構酶抑制劑能特異性導致靶酶失活, 阻斷芳構化反應, 抑制雌激素生成,降低血液中雌激素水平從而達到治療乳腺癌的目的, 並相繼報導了一系列抑制AR 活性的化合物。芳構酶抑制劑作為乳腺癌治療的一種全新思路引起了相關學者的關注, 經近三十年的努力, 已開發上市了三代AR 抑制劑, 作為治療乳腺癌的二線或三線藥物, 在美國、日本等已廣泛用於臨床。AR 抑制劑按其結構可分為甾體和非甾體兩類。具體分類見表1,具體結構見圖1.
 芳香化酶抑制劑
芳香化酶抑制劑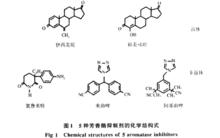 芳香化酶抑制劑
芳香化酶抑制劑2.1 甾體類芳香化酶抑制劑
2.1.1依西美坦(exemestane,Aromasin)
1988 年, 義大利Pharmaela &Upjoin公司研究開發成功新一代抗芳香化酶藥物依西美坦,屬於第三代芳香酶抑制劑 並於1999 年底獲得歐美等國家批准, 主要用於治療絕經後婦女的晚期乳腺癌。
依西美坦特點:①與體內芳香化酶的結合是不可逆的, 實屬芳香化酶滅活劑。依西美坦為芳香化酶的假性底物, 其結構與酶的自然底物雄烯二酮和睪酮相似, 可通過與酶的活性位點不可逆地結合而使其失活(該作用也稱“ 自毀性抑制”), 從而明顯降低絕經婦女血液循環中的雌激素水平。②對芳香化酶的滅活作用無“反彈” 。體外實驗發現, 當從細胞培養液中分別除去依西美坦、氨基導眠能和阿那曲唑後, 依西美坦組JEG-3 細胞中的芳香化酶仍處於受抑狀態, 而後兩組JEG-3細胞的芳香化酶活性反較原來有所提高。套用芳香化酶抑制劑後所致的酶活性提高可能會損害芳香化酶抑制劑的長期療效, 而依西美坦具有的持續抑制芳香化酶活性且不提高芳香化酶蛋白水平的性質, 則有益於減少這種風險。③底物選擇性高, 對腎上腺內皮質類固醇和醛固酮的生物合成無明顯影響。依西美坦即使在高於其抑制芳香化酶作用濃度600倍時, 對類固醇生成途徑中的其他酶也無明顯影響。
2002 年5月, 各國際協作組合作, 選擇絕經後乳腺癌患者, 開展了42 項依西美坦臨床試驗, 內容涉及乳腺癌的預防、術後輔助治療、新輔助治療、轉移性乳腺癌的治療等方面, 以期取得充分的循證醫學依據。
2.1.2福美司坦(formestane)
福美司坦為人胎盤微粒體、大鼠卵巢微粒體、人乳腺癌細胞和乳腺癌活檢樣品中的芳香化酶抑制劑,屬於第二代芳香化酶抑制劑。徐積恩的研究顯示,福美司坦可引起大鼠卵巢和人胎盤微粒體的迅速抑制,給藥後3~20 min 活性抑制可達50%。福美司坦50mg/(kg·d)後採用同位素示蹤法測出雄烯二酮轉化為雌激素的抑制率達90%。
Dowsett M等的研究顯示,給予絕經後女性福美司坦500 mg,24 h 內平均血漿雌激素水平下降40%,給藥7 d 可下降80%;每2 周注射250 mg,8周內抑制雌激素水平約60%;其口服最大有效劑量為250 mg/d。由於絕經前女性雌激素生成主要在腦垂體釋放的促性腺激素控制下的卵巢,不受芳香化酶抑制作用而減少,因而絕經前女性單用福美司坦對雌二醇水平無明顯影響,臨床表現為產生拮抗作用。
Brodie AM等的研究採用二甲基苯並蒽(DMBA)誘發大鼠乳腺癌模型,給予福美司坦50 mg/kg,im,bid,給藥4 周后,約90%的大鼠腫瘤縮小程度>50%,4%的大鼠腫瘤完全消退。Chander SK等的研究顯示,福美司坦12.5 mg/kg+5%檸檬(limonene)對致癌物N-亞硝基脲(NMU)誘發的大鼠乳腺腫瘤有效,抑制率為83.3%,優於福美司坦單獨使用。Dowsett M等發現,採用口服/非胃腸道吸收的所有途徑給藥,福美司坦對血清中的雄烷二醇、睪丸素或5α-二氫睪丸酮水平沒有影響;福美司坦250 mg/d,po,給藥4 次後,34%的患者出現性激素結合球蛋白(SHBG)水平下降,採用氣-質聯用(GC-MS)分析方法測定非胃腸道吸收給藥對血清雌酮水平的抑制約為40%。福美司坦對雌激素生物合成的抑制並不改變絕經後女性的雌激素前體含量。
福美司坦的新用途和用法逐漸被發現,比如對早期子宮內膜癌的治療、對念珠菌的抑制作用、通過與曲妥珠單抗聯用治療乳腺癌、與寬纓酮的聯合抑制芳香化酶作用,增強特異性抗體對腫瘤細胞的抑制作用、降低單一用藥的不良反應等。
2. 2 非甾體芳香化酶抑制劑
2.2.1氨魯米特(aminoglutethimide,AG,氨基導眠能,氨格魯米特,氨苯哌酮)
氨魯米特是一種類固醇生物合成抑制劑。氨魯米特雖是芳香化酶的強效抑制劑, 但對轉化類固醇成為孕烯醇酮的碳鏈裂解酶也有抑制作用, 減少了皮質類固醇的體內合成, 常引起皮疹、共濟失調和磕睡, 發生率高達60 %, 需與氫化可的松合用。偶可出現白細胞減少,血小板減少和甲狀腺功能減退。非甾體芳香化酶抑制劑含有雜原子(通常帶有含N 的雜環部分)。一般通過與細胞色素P 450的血紅素鐵結合, 干擾甾體經基化過程。如同多數甾體類抑制劑一樣,這些化合物是可逆性芳香化酶抑制劑。總體上看, 多數可逆性非甾體芳香酶抑制劑特異性較低, 會不同程度地抑制其它細胞色素P 450 參與的甾體合成的羥基化反應。除氨魯米特外, 其它非甾體芳香酶抑制劑對芳香化酶具有較高的選擇性。
2.2.2來曲唑( letrozole)
來曲唑是高選擇性、第三代芳香化酶抑制劑。它在療效、安全性以及經濟學上的優越性已經為多項臨床研究所證實。來曲唑作為可逆性結合的芳香化酶抑制劑, 通過抑制外周和腫瘤組織中的芳香化酶, 有效降低血漿雌激素水平, 從而去除對激素敏感腫瘤的刺激。約有1/3以上的乳腺癌依賴於雌激素的刺激而繼續發展。在絕經前婦女的雌激素主要來源於卵巢,而絕經後婦女雌激素則主要由腎上腺、脂肪、肌肉、肝臟產生的雄激素經芳香化酶轉化而來。因而對絕經後婦女而言, 通過抑制芳香化酶, 即可減少體內雌激素產生, 從而達到治療乳腺癌的目的。類固醇分子的芳香化是雌激素生物合成過程中的最後步驟, 選擇性芳香化酶抵製劑是能夠作用於雌激素生物合成最終階段的內分泌治療藥物, 不干擾其他固醇類激素的合成, 所以能更好地針對目標, 在作用和療效上有更好的選擇性。
絕經後晚期乳腺癌的一線治療:PO25是1項多中心、隨機、雙盲、雙模擬對照試驗, 旨在比較第三代芳香化酶抑制劑和他莫昔芬作為絕經後乳腺癌一線治療的可行性。此研究涉及29個國家的10個醫療研究機構, 共有916位絕經後晚期乳腺癌入組試驗, 中位隨訪期為32個月。結果來曲唑組的療效均優於他莫昔芬組。
二線解救治療:來曲唑與第一代芳香化酶抑制劑氨基導眠能(AG)的比較:二線研究入組555例他莫昔芬失敗患者, 分別每天服來曲唑2.5mg、0.5 mg和AG 250 mg, 3組有效率分別為19.5%、16.7%和12.4%, 有效和穩定的持續時間分別為21個月、18個月和14個月。來曲唑2.5 mg組的腫瘤進展時間(TTP)、治療失敗時間TTF和總生存均明顯優於AG組,也略優於0.5 mg組。
新輔助治療:研究顯示他莫昔芬治療過的患者發生對側乳腺癌的幾率低於對照組, 由此發現他莫昔芬有預防乳腺癌的作用。但只是阻斷雌激素的作用, 而不能減少其合成, 所以無法去除雌激素代謝產物的潛在致癌作用, 即引起子宮內膜癌變的副作用, 而第三代芳香化酶抑制劑能夠抑制雌激素的合成, 彌補他莫昔芬的不足,理論上預防作用更佳。來曲唑國際協作多中心Ⅲ期臨床研究,絕經後、ER(雌激素受體)和(或)PR(孕激素受體)陽性原髮乳腺癌患者, 162例每天用來曲唑2.5 mg。175例每天他莫昔芬20 mg均為4個月。結果顯示, 每一項研究終點, 來曲唑組均顯著優於他莫西芬組, 有更多的患者適於進行BCS, 45%的患者從保留乳房中獲益。來曲唑組患者客觀緩解率(ORR)顯著高於他莫昔芬組(55% vs36% , P< 0.001), 與他莫昔芬相比, 超過50% 的患者經來曲唑治療後獲益。
通過對目前眾多的大型臨床試驗進行綜合, 證實來曲唑在絕經後乳腺癌治療的各階段, 即在晚期乳腺癌的一線治療、原發性乳腺癌的新輔助治療以及早期乳腺癌的後續強化治療中, 均有卓越的臨床療效, 並且副作用較小, 患者依從性高。在早期乳腺癌的輔助治療中的作用將在目前正在進行的臨床試驗中得出結論。一些基因水平的研究將更加深入的揭示出來曲唑的臨床套用價值、套用特點, 從而更好地指導來曲唑的臨床套用, 使更多的患者從中受益。來曲唑對雌激素具有強烈抑制作用的特點, 也為它開拓其他與女性激素水平有關的癌症治療領域奠定了基礎。
2.2.3阿那曲唑(Anastrozole)
阿那曲唑是第3 代芳香化酶抑制劑, 於1995年經美國食品和藥品管理局(FDA)批准用於治療轉移性乳腺癌。在中國, 從1997 年9月至1998 年6月進行了進口前的臨床驗證研究, 單藥治療絕經後晚期乳腺癌患者, 獲得25.0 %的有效率,於1999 年經中國國家藥品監督管理局(CFDA)批准正式在國內上市。
阿那曲唑適用於經他莫昔芬及其它抗雌激素療法仍不能控制的絕經後婦女的晚期乳腺癌。對雌激素受體陰性的病人,若其對他莫昔芬呈現陽性的臨床反應,可使用阿那曲唑。其可抑制絕經期後患者腎上腺中生成的雄烯二酮轉化為雌酮,從而明顯地降低血漿雌激素水平,產生抑制乳腺腫瘤生長的作用。另外,阿那曲唑對腎上腺皮質類固醇或醛固酮的生成沒有明顯影響。
阿那曲唑用於二線解救治療:大規模臨床研究結果顯示, 阿那曲唑作為一線藥物, 可代替他莫昔芬用於治療絕經後婦女的晚期乳腺癌, 並可考慮為首選一線治療。阿那曲唑具有強效芳香化酶抑制作用, 在外周血循環和腫瘤組織內部均能達到近完全雌激素抑制。1998 年完成的一項多中心Ⅲ期臨床試驗中, 比較阿那曲唑與甲地孕酮治療既往他莫昔芬失敗患者。兩種藥物取得相似的臨床獲益率, 阿那曲唑1mg組的中位死亡時間為26 .7 個月, 甲地孕酮組為22 .5 個月。阿那曲唑治療組在生存率方面明顯優於甲地孕酮組。研究結果也顯示阿那曲唑治療晚期乳腺癌的耐受性比甲地孕酮更好。因此阿那曲唑作為二線解救治療可以取代甲地孕酮。
阿那曲唑用於輔助治療:阿那曲唑在一線治療中的數據顯示了阿那曲唑在早期乳腺癌治療中的地位。因此又有阿那曲唑、他莫昔芬單獨和聯合套用試驗(Anast rozole Tamox ifenAlone or Combination , ATAC 試驗)研究, 目的通過輔助治療中與他莫昔芬比較以證實其在早期乳腺癌患者中具有同樣的療效。
阿那曲唑與其他第3 代芳香化酶抑制劑在解救治療中的對比研究:目前有代表性的第3 代芳香化酶抑制劑主要是甾體類的依西美坦和非甾體類的阿那曲唑、來曲唑。在19個國家、110箇中心進行了一項大型研究, 比較來曲唑和阿那曲唑兩藥用於二線治療絕經後晚期乳腺癌。研究共有713例患者入組。來曲唑組356 例, 每日口服來曲唑2.5mg ;阿那曲唑組357例,每日口服阿那曲唑1mg。兩組病例均為激素受體陽性或受體不明者。研究中每3個月評價療效。研究結果顯示, 來曲唑組的總反應率(OR)較阿那曲唑組高,分別為19 .1 %和12 .3 %(P =0 .014)。來曲唑的臨床獲益率數值較阿那曲唑組高, 分別為27 %和23 %, 但無統計學差異(P =0 .218)。而兩藥的腫瘤進展時間TTP、治療失敗時(TTF)、反應持續時間和臨床獲益時間上無差別。研究顯示兩藥的耐受性良好。
阿那曲唑單藥顯示出良好的療效及耐受性, 它已經在乳腺癌的一線、二線治療,輔助治療、新輔助治療中全面超越傳統藥物他莫昔芬,成為可能替代他莫昔芬的理想藥物。
3.中國指南關於芳香酶抑制劑的使用
3.1對於沒有接受過抗雌激素治療或無復發時間較長的絕經後復發患者,他莫昔芬、芳香化酶抑制劑或氟維司群都是合理的選擇。絕經後復發轉移性乳腺癌,一線內分泌治療的首選為第3代芳香化酶抑制劑,包括阿那曲唑、來曲唑、依西美坦,因為在他莫昔芬治療失敗的復發轉移性乳腺癌的二線治療中,第3代芳香化酶抑制劑比甲地孕酮更有效。在復發轉移性乳腺癌的一線內分泌治療中,第三代的芳香化酶抑制劑明顯優於他莫昔芬。絕經前復發轉移性乳腺癌患者首選化療,如果激素受體陽性患者適合或需要用芳香化酶抑制劑進行內分泌治療時,首選雙側卵巢切除手術,後續聯合芳香化酶抑制劑。藥物性卵巢功能抑制聯合芳香化酶抑制劑也是可以考慮的方案(但尚缺乏臨床證據)。
3.2絕經後的患者,一線內分泌治療可以選擇芳香化酶抑制、氟維司群、他莫昔芬或托瑞米芬。通常會優先選擇芳香化酶抑制劑,存在芳香化酶抑制劑治療禁忌證、曾行芳香化酶抑制劑輔助內分泌治療且無病生存時間短、或因經濟原因不能接受芳香化酶抑制劑治療的患者,可考慮給予他莫昔芬或托瑞米芬。
3.3可以換用另一類芳香化酶抑制劑。如非甾體類芳香化酶抑制劑(來曲唑、阿那曲唑)治療失敗後,可以考慮換為甾體類芳香化酶抑制依西美坦治療,反之亦然。
