產生背景
隨著中國2010版GMP的順利頒布,無菌藥品、風險和驗證,再次成為製藥行業內討論的熱點。除菌過濾工藝的風險和驗證,也因為國外法規和新版GMP對其的關注,成為了業內普遍關注的問題。
除菌過濾工藝
除菌過濾 是指在不影響產品質量的前提下,過濾去除流體中微生物的工藝過程。以液體過濾為例,雖然符合上述定義描述的工藝過程在藥品生產中有著廣泛和普遍的套用,但是,我們需要區別下述兩種不同的情形:第一種是指因為產品、中間體或者其他過程料液具有不穩定性,不能採用包括熱滅菌在內的被監管部門認可的最終滅菌方法,而採用過濾除菌的方法,並要求濾出液無菌的情況;第二種則是指採用具有不同精度(pore size rating)的過濾器,甚至是除菌過濾器,對工藝流體進行過濾,而對濾出液只有微生物污染水平的要求,並沒有無菌要求的情況。
上述二者中,前者可以被理解為是對除菌過濾的狹義定義,簡稱“除菌過濾”;而後者是更廣義的概念,通常被稱為“微生物污染水平(bioburden)控制過濾”,該過程中使用的過濾器被稱為“微生物污染水平控制過濾器”,以區別於第一種情況。在不同套用中,法規對過濾器的驗證要求,也有區別的。
除菌過濾器
各國監管機構對除菌過濾器的定義都已經有了統一的認識,並被寫入法規要求或指南中。
美國ASTM F-838標準現在已經成為確認和驗證液體除菌級過濾器微生物截留效能的標準方法,即每平方厘米有效過濾面積挑戰水平達到107 CFU,挑戰微生物為缺陷假單胞菌(Brevundimonas diminuta, ATCC 19146)的情況下,濾出液無菌的過濾器。
目前市場上,除菌過濾器通常被標示為0.2μm或者0.22μm孔徑精度,但是,應當注意兩點,即:這兩種精度是可以互換的,沒有區別;非除菌級的過濾器產品,也有標示為0.2μm(或者0.22μm)精度的。因而,選擇和判斷過濾器是否是“除菌級”的,不能僅根據其孔徑是否為0.2μm(或者0.22μm)精度,還需要根據上述法規中的定義進行。
驗證概要
採用除菌過濾工藝的無菌藥品生產中,過濾所需時間、濾出總量、過濾器兩側壓差以及重複使用等,都會影響除菌過濾器的細菌截留能力。為確保藥物安全及生產的穩定性和重複性,按照《藥品生產質量管理規範(2010年修訂)》(簡稱新版藥品GMP) 附錄Ⅰ《無菌藥品》中的規定,需要對除菌過濾器進行驗證。
美國FDA Guidance for Industry – Sterile Drug ProductsProduced by AsepticProcessing,歐盟GMP Annex 1 SterileMedicinal Products,美國注射劑協會PDA Technical Report # 26Sterilizing Filtration ofLiquids等,對除菌過濾器驗證也都有明確的驗證要求。
驗證責任劃分
按照GMP藥品實施指南的建議,除菌過濾器的驗證一般由過濾器生產商提供,使用者進行確認。
驗證內容
《標準驗證指南-StandardValidation Guide》: 過濾器的使用者從過濾器的生產商處獲取的證明檔案,證明其使用的過濾器適用於在製藥過程中套用。該檔案一般為採用法定的方法檢測過濾器後的數據證明, 支持但是不能代替過濾器的性能驗證。
1.《標準驗證指南-StandardValidation Guide》: 過濾器的使用者從過濾器的生產商處獲取的證明檔案,證明其使用的過濾器適用於在製藥過程中套用。該檔案一般為採用法定的方法檢測過濾器後的數據證明, 支持但是不能代替過濾器的性能驗證。
 除菌過濾器驗證
除菌過濾器驗證2、除菌過濾器在工藝條件下的再驗證,性能驗證。
工藝條件下再驗證
由過濾器生產商提供除菌過濾器在工藝條件下的再驗證,是法規認可的。
例如某除菌過濾器供應商提供的驗證內容,包括但不僅限於:
化學兼容性、溶出物試驗、細菌存活試驗、細菌挑戰試驗、吸附試驗、產品潤濕的完整性測試參數及重複使用驗證等。
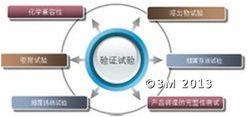 驗證實驗
驗證實驗驗證方法
試驗條件:採用與實際工況相同或者風險更高的條件
 試驗條件
試驗條件1. 化學兼容性:外觀檢查、稱重、完整性測試
2. 溶出物試驗:通常採用替代溶液,對溶出物進行紅外、GC-MS、或TOC檢測對照除菌過濾器的標準圖譜。這些測試方法無優先權。
3. 細菌存活試驗:將缺陷型假單胞菌(ATCC19146)接種到料液中,接觸時間達到或者超過最長工藝時間,取樣培養,判斷菌落數是否超過1個Log的下降。
如果有,則判斷為料液是抑菌的,需採用替代溶液或者改變測試條件(接觸時間、藥液濃度等);將挑戰菌接種到替代溶液或者藥液中進行下一步的挑戰實驗。
如果沒有,則判斷為無抑菌,可以將挑戰菌直接接種到藥液中。
4.細菌挑戰試驗:挑戰菌缺陷型假單胞菌(ATCC19146),挑戰濃度高於10cfu/cm過濾面積,進行多組平行試驗,其中一組過濾器的起泡點接近臨界值。
5. 吸附試驗:通常在客戶端進行,過濾一定量料液後,檢測過濾後料液中的有效成分含量。
6. 產品潤濕的完整性測試參數:用多組濾膜,其中一組的起泡點接近臨界值,首先用標準方法檢測試完整性。乾燥後,用料液潤濕再檢測完整性。每組重複三次。計算得出,產品潤濕的完整性測試參數。
實驗室資質
提供除菌過濾器驗證服務的實驗室,通常需具備ISO 9001, ISO17025以及CNAS認證。
 實驗室 實驗室 |  實驗室 實驗室 |  實驗室 實驗室 |
